摘 要
目的:
分析抗肿瘤药物临床试验的质量控制因素,找出抗肿瘤药物临床试验中的常见问题,并提出相应的解决对策,为相关单位提供科学有效的依据。
方法:
研究采用的主要方法是文献分析法及实地问卷调研法。笔者在论文选题、撰写、调查问卷设计的过程中查阅了大量的文献。笔者对国内四家有肿瘤科药物临床试验资质的 100 名肿瘤专科医院研究者进行了实地问卷调研,以分析抗肿瘤药物临床试验的质量控制因素,找出抗肿瘤药物临床试验中的常见问题。该调查问卷的克伦巴赫系数为 0.901,说明问卷的信度较高,可靠性也较高;KMO (Kaiser-Meyer-Olkin) 值为0.803,说明问卷的结构效度较高。问卷回收后的数据用 Epidata3.1 及 SPSS20.0 软件进行数据录入、整理、校对、分析。数据分析方法为因子分析法。
结果:
通过对国内外药物临床试验相关法规的对比分析,发现由于我国药物临床试验起步较晚,目前国内 GCP 与国际 ICH-GCP 相比,在内容的完整性及先进性上仍然存在着一定差距。对调查对象的性别、学历及职称进行了评估,发现参与药物临床试验的女性研究者较多,总体研究者以硕士及以上学历的居多,大多参与临床试验的研究者是中级职称。因此,抗肿瘤药物临床试验的研究者大多学历较高,具有丰富的临床经验。通过将收集回来的数据进行因子分析,发现抗肿瘤药物临床试验的质量控制因素是:研究者对临床试验的管理、对受试者的管理、对临床试验数据的管理、对研究药物的管理、伦理委员会对临床试验的管理、药物临床试验机构对临床试验的管理、对中心实验室样本的管理。抗肿瘤药物临床试验中的常见问题是:研究者对临床试验的管理力度不够、对受试者管理的欠缺、临床试验数据的管理不规范、研究药物的管理不规范、伦理委员会对临床试验的管理力度不够、药物临床试验机构对临床试验的管理力度不够、对中心实验室样本的管理不规范。
结论:
在了解我国抗肿瘤药物临床试验中的质量控制因素及常见问题的基础上,为提高我国抗肿瘤药物临床试验的质量,提出了以下建议:提高研究者对临床试验的管理力度、加强对受试者的管理、加强对临床试验数据的管理、规范研究药物的管理、提升伦理委员会对临床试验的管理力度、提升药物临床试验机构对临床试验的管理力度、规范中心实验室样本的管理。
关键词:抗肿瘤,临床试验,质量控制,因素
Abstract
Objective: To study the clinical effect of the method
Objective to analyze the quality control factors of clinical trials of antineoplastic drugs, find out the common problems in clinical trials of anti-tumor drugs, and put forward the corresponding countermeasures, so as to provide scientific and effective basis for relevant units.
method:
The main research methods are literature analysis and field survey. The author consulted a lot of literature in the process of topic selection, writing and questionnaire design. In order to analyze the quality control factors of clinical trials of antineoplastic drugs and find out the common problems in the clinical trials of anti-tumor drugs, the author conducted a field survey on 100 researchers in four cancer hospitals with the qualification of clinical trials of oncology. The Cronbach coefficient of the questionnaire is 0.901, indicating that the reliability and reliability of the questionnaire are high; the kmo (Kaiser Meyer Olkin) value is 0.803, indicating that the structure validity of the questionnaire is high. The collected data were input, collated, checked and analyzed by epidata3.1 and spss20.0 software. The data analysis method is factor analysis.
result:
Through comparative analysis of the relevant laws and regulations in domestic and foreign drug clinical trials, it is found that due to the late start of clinical trials in China, there is still a certain gap between the domestic GCP and the international ICH-GCP in terms of completeness and advanced nature of the contents. The gender, educational background and professional title of the subjects were evaluated. It was found that there were more female researchers participating in drug clinical trials. The majority of the researchers had master's degree or above, and most of the researchers participated in clinical trials were intermediate titles. Therefore, most of the researchers in clinical trials of anti-tumor drugs have higher education and rich clinical experience. Through factor analysis of the collected data, it is found that the quality control factors of clinical trials of antitumor drugs are: the management of clinical trials by researchers, the management of subjects, the management of clinical trial data, the management of research drugs, the management of clinical trials by ethics committee, the management of clinical trials by drug clinical trial institutions, and the sample of central laboratory Management. The common problems in clinical trials of antineoplastic drugs are: the management of clinical trials by researchers is not enough, the management of subjects is not enough, the management of clinical trial data is not standardized, the management of research drugs is not standardized, the management of clinical trials by ethics committee is not enough, the management of clinical trials by drug clinical trial institutions is not enough, and the management of central laboratory samples is not enough The reason is not standard.
Conclusion
On the basis of understanding the quality control factors and common problems in the clinical trials of anti-tumor drugs in China, the following suggestions are put forward to improve the quality of clinical trials of anti-tumor drugs in China: to improve the management of researchers on clinical trials, to strengthen the management of subjects, to strengthen the management of clinical trial data, to standardize the management of research drugs, and to improve the quality of clinical trials of anti-tumor drugs To improve the management of clinical trials in drug clinical trial institutions and standardize the management of central laboratory samples.
Key words: antitumor, clinical trials, quality control, factors
前 言
恶性肿瘤严重威胁了人类的生命健康。虽然现有的治疗手段目前已经取得了一定的疗效,但由于大多数的恶性肿瘤患者缺乏有效的治疗药物,其生存时间有限,开发新的抗肿瘤药物成为了人类的新课题。目前,抗肿瘤药物的临床试验是国际上投资最大、投入最多的领域。
因而,各个国家的政府、机构和制药公司对抗肿瘤药物及相关的抗肿瘤研究都给予了高度的重视。近年以来,随着医疗科技对肿瘤相关机制的逐渐阐明,分子药理学及分子肿瘤学的不断发展,基因工程、大规模快速筛选、组合化学等先进技术的进一步应用,加快了抗肿瘤药物的开发。
1983 年,我国卫生部门首次建立了十四家临床试验基地,这成为了我国药物临床试验的开端。在全球医药一体化的推动下,药物临床试验在我国逐渐形成市场化、产业化的模式。因为在我国的临床试验研究总体费用投入较低,病种及病源分布广泛,人口资源丰富,这使得我国逐渐成为药物临床试验行业的关注焦点[1].我国目前仅有六千多家医药相关企业投入到药物临床试验的行业中,总体研发能力及市场的总额在全球市场占比小于百分之十。但是,我国有信心,也有能力将药物临床试验做得更好,为我国人民提供更多的新型治疗方案及高质量的新型药物。
我国的药物临床试验历经多年的发展得到了令人瞩目的成绩,但也显现出较多问题,例如临床试验过程中规范性不够、受试者依从性较差、研究者的 GCP 意识不足、相关人员的培训不够、质量监控体系、监督力度需要加强等等。
本课题旨在通过对相关因素的论证和比较、综合分析,研究和总结药物临床试验中的常见问题,进一步明确我国抗肿瘤药物临床试验质量存在的问题,探索符合中国国情的改进措施[2],为提高研究中心抗肿瘤药物临床试验的质量、完善我国抗肿瘤药物临床试验的质量监管体系提供了有效建议。
1 国内外药物临床试验发展现状分析
1.1 国外药物临床试验发展现状
最早的外国临床试验记录大约在公元前 600 年,当时巴比伦国王尼布甲尼撒二世进行了饮食测试。测试人群被分为两组,其中一组仅吃蔬菜,另一组仅吃肉,最后得出了只吃蔬菜的那一组人群更有生气的结论[3].后续人类历史上渐渐出现以治疗人类疾病为目的的临床试验,例如 1747 年 James Lind 医生进行的第一个"有对照组的临床试验".该试验采用不同的治疗方法将 12 位坏血症患者分为 6 组,一组病人每天吃柠檬和橘子,其他病人每天吃苹果酒、醋、海水等。最终发现使用柠檬和橘子的那一组患者痊愈,得出"坏血病的主要病因是缺乏维生素 C"的结论[4].
统计学专家布兰德福德于 1950 年进行了一项用链霉素治疗肺结核的研究,用于观察其有效性。这是第一次有记录的随机分组及双盲对照研究,用统一的分析指标由独立在该试验外的相关医务人员进行评估。该试验也被认为是近代首个随机药物临床试验[5].
上个世纪八十年代的中期,全球三十多个国家,共计上千家医院建立了研究合作中心,即国际性协作组,以解决前期出现过的临床试验的样本量过少的问题。在短短两年的时间里,研究中心共同入组了六万多名受试者,开创了临床试验中心国际性合作的研究方式。这种方式为增加受试者入组、提高临床试验的效率起到了极为重要的作用[6].
随着时间慢慢推移,人类医疗保健水平进一步提高。在临床试验一项项进行的过程中,很多新型药物慢慢被开发出来,出现了抗抑郁药物、抗肿瘤药物、抗菌药物等新药,大大提高了人类的生存质量。然而,当时对试验药物的有效性和安全性的评价是不充分的,并且也受到了临床试验手段的限制,出现过上市药品不能发挥其预期疗效或者在获得疗效的时候受到严重的身体损害,甚至导致患者死亡的情况。也因为对药物临床试验缺乏有效监管,发生过某些研究者滥用受试者进行临床试验以了解药物的机制及功能的情况。例如利用孕妇、残疾人及囚犯进行放射物试验,对于患者在未经任何治疗的情况下进行长达 40 年的后续研究,对受试者的健康和生命造成严重损害。为了保证药物临床试验的规范性,保护受试者的基本权益,为新药上市能够提供可靠、准确的临床试验数据,各个国家的相关部门开始建立监管药物临床试验的体系[7].国外监管药物临床试验的体系发展史可分为三个阶段[8]:
(1)第一阶段(20 世纪初-20 世纪 60 年代):监管药物临床试验的体系初步形成美国在 1937 年发生过轰动一时的磺胺酏剂事件。当时 Harold Watkins 担任Massengill 公司的药剂师,他用二甘醇替换乙醇将磺胺溶解,用来制作成方便儿童服用、口感较好的口服液,并且将该口服液命名成"磺胺酏剂".这批药物没有经过临床试验就销往市场,造成 300 多人肾功能衰竭以及 107 人中毒死亡的严重后果。后来进行动物实验,证明造成死亡的原因是工业用的二甘醇,磺胺实际无毒性。当时由于相关法律并没有规定药物必须要经过试验才能进行售卖,联邦法院只能判定Massengill 公司为"掺假及贴假标签",并该公司处以 26100 美金罚款[9].最终哈罗德·沃特金斯在绝望和内疚中自杀,但该事件已经对患者造成了无法弥补的后果。该事件促使美国当局意识到药物在上市前进行临床试验的重要性,由当时的总统罗斯福于 1938 年签署《食品、药物与化妆品法案》,替代 1906 年的《纯食品和药品法》。
该法案规定新的药物上市之前必须将证明药物安全性的临床试验相关资料提供给监管部门。随后,美国国立卫生研究院于 1953 年颁布了一项联邦法令,要求必须在临床试验开始前进行独立审查[7].
"磺胺酏剂"事件发生之后,规定新药一定要进行动物毒性相关试验,但对于试验的样本数量、观察指标等标准没有确立明确的要求。另外,第二次世界大战发生之后,胰岛素、抗生素等医疗研究进入异常迅速发展的时代。随之而来的,是新的医疗事件发生:(1)黄体酮早已在动物试验上发现毒性,1939 年发现化学合成孕激素的结构疑似男性激素,会导致后代的雌性动物雄性化。但由于当时人们并没有重视这个问题,反而在 1937 至 1959 年之间,将其用于治疗美国妇女的先兆性流产,导致六百多名新生女婴生殖器男性化;(2)法国有机锡胶囊的急性毒性试验观察时间不足,本应观察 3-7 天,实际仅观察了 24 小时,因此半数致死量的计算结果并不准确。最终导致在 1954 至 1956 年之间,207 人视力出现障碍,其中 102 人甚至死亡[10].
随后在 1956 年,原联邦德国的格伦南苏制药厂研发生产了治疗妊娠呕吐的镇静药,名为沙利度胺,又称为反应停。在药物上市的 6 年时间里,其临床疗效非常明显,被澳大利亚、原联邦德国、加拿大、拉丁美洲、非洲等共计 28 个国家及地区广泛地使用。直到 1960 年,各国陆续发现新生儿的四肢呈"海豹肢畸形"的几率增加,相关动物实验和流行病学调查也证明"海豹肢畸形"是因为孕妇服用沙利度胺引起。该药物未严格经过临床前的实验,在售卖过程中收到过一百多例毒性反应相关的报告,但均被隐瞒下来。最后导致日本有一千多名"海豹肢畸形"的儿童,德国大概有八千多名,全世界超过一万名受害者。此事件成为轰动一时的"反应停事件"[10].
上述事件让全世界对新药在上市前进行安全性审查的必要性进行了深入思考,许多国家认识到必须在新药上市前进行安全性评估,于是纷纷开始设立相关部门进行监管。这一事件促使美国在 1962 年通过法案《Coff-Waharis Amendment》,对《食品,药品和化妆品法案》进行了重大修订。法案要求每个制药公司必须向 FDA 提供与临床试验相关的信息,以在新药上市之前证明药物的有效性和安全性。另外,制药公司必须保存所有药物不良反应的相关资料[7].此外,该法案明确规定了新药上市前的批准程序,并明确了新药上市的申请要求和药物临床试验的申请要求[11].
在 20 世纪 30 至 60 年代之间,世界范围内还发生过多例恶意侵犯受试者权益、违背医学伦理的事件。位于美国纽约的 Willowbrook 州立学校是一家专门收留智障儿的学校。该学校 1956 年规定,参与肝炎研究的儿童可以直接上学而不必排队,否则他们需要排队等候两年才能重新入学。这项规定导致 14 年内,百分之八十五的儿童,共计七百多名患上了肝炎。
美国纽约的犹太人慢性病医院于 1963 年在没有取得 21 名意识不清、身体状况差的患者知情同意的情况下,仅告知病人是进行皮试,实际向病人注射肝癌细胞,用来观察人体器官的移植排斥过程[7].
美国公共卫生部自 1932 年在六百多名非洲裔美国贫困男性中,进行了一项名为Tuskegee 的梅毒研究,该研究持续了 40 年。在研究过程中,研究人员隐瞒了梅毒携带者的真实情况,并且在发现有效治疗时未对患者进行任何治疗,以此来观察梅毒的发展过程[12].在该试验被揭露后,美国此后一段时间内颁布了一系列和临床试验中受试者保护相关的法规。
(2)第二阶段(20 世纪 70 年代-20 世纪 80 年代):全球药物临床试验逐渐形成规范化及法制化的监管体系随着医疗科技的逐步发展,保障受试者的权益渐渐成为国际性的共识,医学伦理被认为是临床试验开展的前提。药物临床试验不仅要在数据上严谨可靠,更需要在伦理上获得认同。为使临床试验的伦理规范得到统一,芬兰赫尔辛基于 1964 年召开了第十八届世界医学大会,会议上通过了《赫尔辛基宣言》。这个宣言在保留《纽伦堡法典》基本精神的情况下,对受试者权利的保护进行了更严格的要求,声明医生最首要的职责是让受试者的健康和生命得到保障,制定了人体对象医学研究实验的伦理规范及道德准则。该宣言也可被视为 GCP 的初始雏形[7].
美国联邦福利、教育与卫生部于 1974 年制订并推广了《美国国家研究法》,将受试者保护的相关要求纳入法规之中。在同一年,美国国会也设立了"保护生物医学与行为学研究中的受试者委员会"[13],并于 1979 年发布了《贝尔蒙特报告》的历史性文件。该报告确定了所有与人类相关的研究应遵循的基本原则,即善行,尊重和公正[14].1982 年颁布了和生物医学研究的人类受试者相关的《国际伦理指南》,该指南规定了发展中国家开展生物医学相关研究的伦理标准[15].
(3)第三阶段(20 世纪 90 年代至今):全球药物临床试验统一标准逐渐形成二十世纪八十年代末至二十世纪九十年代初期,美国在深入探讨临床试验规范后,制定了 GCP,即药物临床试验管理规范。该规范被纳入到国家法律体系中,成为了当时全球药物临床试验监管的最高标准。后来日本、韩国、欧共体、加拿大、澳大利亚等各国都在总结经验教训的基础上,陆续颁布并实施了符合各个国情的临床试验管理规范[15].世界卫生组织总结了各国 GCP 的相关内容后,于 1993 年草拟了《药物临床试验规范指导原则》[16].
药物临床试验管理的一般原则在所有国家和地区都是大致相同的,但具体的细节标准不同。因此,来自一个国家或地区的临床试验的数据通常不被另一个国家或地区承认。在这样的背景下,同一个企业的同一种产品在一个国家被批准上市后,还需要根据不同的标准在其他地区和国家重复相同的临床试验,从而产生大量的物质资源、人力及财力的浪费。此时,多国政府都意识到了将各国的药物临床试验规范统一进行调试的必要性。于是,国际人用药品注册技术要求协调会(ICH)在人们的热切盼望中成立了。ICH 主要由日本、美国、欧盟的行业组织及药监部门组成。日本于 1996年召开了 ICH 会议,颁布了 ICH-GCP.后续美国、日本、欧盟纷纷开始正式实施ICH-GCP,号召所有药物临床试验应当按照 ICH-GCP 的标准进行质量控制,世界卫生组织、瑞士、澳大利亚、加拿大等国家及地区都表示认可[17].此时,ICH-GCP 开始成为最广泛用于控制药物临床试验质量的标准,药物临床试验的监管体系走入了国际标准统一时期[7].
国际临床数据交换标准协会(CDISC)自 1997 年开始发展全球化独立的数据标准平台,让全球的电子信息系统能够互相兼容,改善了卫生保健和医药研究相关领域的交流和药政监督。目前,CDISC 已经主导了国际试验临床数据的标准化,成为 ICH指导下的全球临床试验研究模式标准。日本和欧美已明确表示过如果药政需要申请数据,必须符合 CDISC 的标准才能申请[18].
《欧盟临床试验指导》于 2001 年颁布。该指导的主要目的是协调和简化欧盟医药临床试验相关的批准程序和管理规定,使受试者的权利和需求在临床试验中得到充分的保护,为欧盟的人体临床试验提供框架性和法律性的文件[18].
1.2 国内药物临床试验发展现状
我国最早的医疗体系以中医及中药为主,一直到近代才开始接受西方的西药及西医治疗体系和方法。根据历史资料记载,有关中国临床试验的最早信息记录在《史记·三皇本记》之"神农尝百草,始有医药"这一个部分中。神农氏以自己为试验对象,亲自品尝百草来观察药物的疗效,他被认为是中国第一个进行药物临床试验的人[19].新中国成立后,中国也出现了以自己为试验对象进行临床试验的医务工作者。
例如汤飞凡(中国第一代的医学病毒学家)于 1957 年将沙眼衣原体接种至自己的眼睛里,造成典型的沙眼。为了观察全部病程,一直坚持四十多天后才去治疗,证明了沙眼衣原体对于人类有致病性[12].
建国后,我国医疗卫生水平快速发展,对医疗相关法规建设的要求越来越高。国家卫生部门根据我国的实际情况,借鉴了国外先进的管理方法和管理理念,稳步建立了具有中国特色药物临床试验的质量控制体系[7].
我国的 GCP 发展大概经历了十多年时间,其制定大致可以划分为两个阶段[12]:
(1)第一阶段(1986-1994 年):信息收集阶段自 1986 年以来,中国一直关注国际 GCP 的发展,从 1993 年开始收集、整理各国的 GCP 法规作为参考资料,并邀请国外的专家来华进行介绍及指导。
(2)第二阶段(1995-1997 年):GCP 草拟及定稿阶段我国 5 位教授李家泰、汪复、诸骇仁、桑国卫、游凯于 1995 年以 ICH-GCP 为基本准则,结合我国实际情况草拟了中国第一个 GCP.初稿完成后,经过了两次修改,在同年 11 月再次提交国家相关部门,形成了《药品临床试验管理规范送审稿》[20],卫生部后于 1998 年 3 月 2 日发布《药品临床试验管理规范(试行)》。当时的国家药品监督管理局在 1998 年重新修订了该文件,1999年9月1 日正式颁布了《药品临床试验管理规范》(GCP)。
国家药品监督管理局于 1999年5月1 日正式颁布了《新生物制品审批办法》 、《进口药品管理办法》等相关法规。同时,国家对 5 个 GCP 临床试验中心进行了重点支持,在全国率先开展 GCP 临床试验工作,使临床试验水平逐步向国际水平靠拢[21].
国际多中心的临床试验由于其试验设计水平在每个国家大致相同,减少了不必要的重复临床试验,获取的是全球的临床试验数据,这缩短了药品研发上市的时间,为各个地区的药物批准提供了更好的数据。因此,目前药物临床研究有着全球化的趋势,国际多中心的临床试验越来越受到青睐[22].中国最早的国际多中心临床试验在 20 世纪 90 年代开展,包括一些跨国制药公司作为申办方在中国开展的国际多中心药物临床试验及与世界卫生组织合作的临床试验项目。国家药品监督管理局于 2002 年发布了针对国际多中心临床试验管理的具体规定之后[1],越来越多国际多中心的临床试验选择在中国开展。据药品审评中心统计,2002 年在我国仅申报了 3 个国际多中心临床试验,但到了 2013 年,数量升至 272 个[1],并且出现逐年上升的趋势[23].
2015 年我国药监部门进行了改革,颁布了与国际多中心临床试验相关的一系列指南,这进一步地推动及鼓励了国际多中心临床试验在我国的发展。药品审评中心于2013 年 9 月要求所有的临床试验必须进行登记。截至到 2018 年 5 月,药品审评中心的登记公示平台上显示国际多中心的临床试验数量已经达到了 748 个。目前我国的国际多中心临床试验大多由国外药企发起,由中国发起的国际多中心临床试验数量屈指可数。自 2002 年起,我国相关部门正式开始对国际多中心临床试验进行监管。从 2015开始,我国政府开始鼓励创新医疗器械及药物尽早进入中国,鼓励国内和国外同步进行临床试验,对于国际多中心临床试验的诸多限制进行放开,使百姓获益[23].
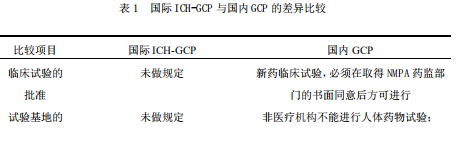
1.3 国际 ICH-GCP 与国内 GCP 的差异比较
国内的 GCP 是根据 ICH-GCP 相关内容,根据国内实际情况结合而成的,二者的基本原则一致。通过二者的对比,会发现 ICH-GCP 与 GCP 在申办方职责、研究者职责、监查员职责、研究中心的资质、临床试验资料保存等方面会有部分差异。我国药物临床试验起步较晚,目前国内 GCP 与国际 ICH-GCP 相比,在内容的完整性及先进性上仍然存在着一定差距。

参考文献
[1] 洪丹,徐艳,李飞燕等。文盲患者参加抗肿瘤药物临床试验的若干伦理问题[J]. 中国肿瘤,2015, 24(10): 838~840.
[2] 黄健,王晓稼。 抗肿瘤药物临床试验中的医学伦理问题[J].中国肿瘤, 2015, 24 (10):834~ 837.
[3] 李洁,廖红舞。 肿瘤临床治疗与研究者中的德行伦理与规范伦理的思考[J]. 医学与哲学,2017,38(3A): 43~44.
[4] 石远凯,孙燕。 中国抗肿瘤新药临床试验研究的历史、现状和未来[J]. 中华医学杂志,2015,95(2): 81~85.
[5] 吴桓兴,周际昌,孙燕等。N-甲酰溶肉瘤素治疗恶性肿瘤初步临床报告[J].中华医学杂志,1962,48(8): 488~494.
[6] 唐永刚。 抗肿瘤药物临床试验现状与研究进展[J]. 药学传论杂志,2008,17(7): 1~3.
[7] 汤致强,戴媛媛。 抗肿瘤药物临床研究进展[J].专家评说,2005, 3(2):5~7.
[8] Osada S, Saji S. New approach to cancer therapy: the application of signal transductionto anti-cancer drug [J]. Curr Med Chem Anti-cancer Agents, 2003, 3(2): 119~131.
[9] Chong H, Vile R. Gene therapy for cancer [J]. Drugs Future, 1997, 2: 857.
[10] Baselga J. New horizons: gene therapy for cancer [J]. Anti - Cancer Drugs, 1999, 10:S39.
[11] 漆璐,王瑜,王兴河。 抗肿瘤药物临床试验中风险评估与应对措施的探讨[J]. 中国临床药理学杂志,2019, 35(10):1058~1060.
[12] 张红梅,孔胜男,王筱雯,张琼,余智操。 抗肿瘤药物临床研究之得与失[J]. 医学与哲学,2018, 35(6B):4~6.
[13] 张雷, 郝纯毅, 廖红舞, 陆婷, 周顺连, 李洁。 抗肿瘤药物临床试验的特点和医学伦理问题[J]. 肿瘤防治研究,2017, 44(7):506~508.
[14] 漆璐,王瑜,雷春璞,王兴河。 抗肿瘤药物临床试验质量管理探讨[J]. 中国临床药理学杂志,2018, 34(12):1479~1488.
[15] 胡薏慧,汤洁,彭朋,元唯安。 关于当前我国新药临床试验质量问题的思考[J]. 广东药科大学学报,2019, 35(2): 279~284.
[16] 谢广宽。 医学研究者的经济利益冲突对临床试验的影响--美国的经验与启示[J].
医学与哲学: 人文社会医学版,2005, 26(10):42~45.
[17] 蒋葵,张阳。 肿瘤药品临床试验中的知情同意[J]. 医学与哲学(临床决策论坛版),2007,28(11): 68~69.
[18] 孙斌。 知情同意为什么难以实现[J]. 医学与哲学,2005, 26(2):22~ 23, 34.
[19] 张馨心,刘锦钰,杨婕,刘宁,王梅红。 人体药物临床试验受试者合法权益保护法律问题研究[J]. 中国卫生法制,2019,27(3): 8~11.
[20] 张同梅,李宝兰。 药物临床试验中如何有效签署知情同意[J]. 临床研究,2019,27(5): 191~193.
[21] 谢振伟,范华莹,王瓅珏,张华,王豪。 药物临床试验数据核查常见问题与对策建议[J]. 中国临床药理学杂志, 2017,33(22): 2299~2307.
[22] 刘龙,漆璐,王进,王瑜,王兴河。 抗肿瘤药物临床试验中不良事件规范化判断的探讨[J]. 中国临床药理学杂志,2019, 35(4):396~398.
[23] 马珂,俞佳。医院药师在药物临床试验中的作用[J]. 中国药房杂志,2005,16(17) :1352~1353.
[24] 罗翠霞,梁小帆。临床试验标准操作规程之药房管理[J]. 实用药物与临床,2009,12(4) : 302~303.
[25] 寇莹莹,冯继锋。 抗肿瘤药物临床试验药品的科学化管理及其意义[J]. 临床合理用药,2015,8(1): 18~19
[26] 丁铃。 抗肿瘤药物临床试验药品的科学化管理探讨[J]. 世界最新医学信息文摘,2017, 17(85):197.
[27] 程雅倩,何文。 我国药物临床试验的开展和监管体系现状[J]. 中国药师,2019,22(6): 1132~1138.
致 谢
回首三年的研究生岁月,有欢笑也有失意,但更多的是不舍。在临近毕业之际,我感慨万千。这三年的求学生涯是我人生中一段重要的经历,在这里我遇到了许多给予过我帮助的人,也学到了很多东西。因此,我想衷心的对你们说声感谢,同时也希望给予你们由衷的祝福。
首先要感谢我的导师章佳安老师。在他的悉心指导及热心鼓励之下,我的论文从选题,到中期报告,再到如今定稿答辩,都得以顺利完成。在与章老师相处的这三年里,他对学问一丝不苟,对学生孜孜不倦,认真严谨的态度时刻影响着我,使我终生受益。
吴诗老师作为我们的班主任,在生活及学习上给予我们真诚的关心及关怀,让我除了能够扎实地学习专业知识,也能感受到家庭般的温暖。
感谢研究生处夏斌老师对于我学习及生活上的帮助、支持和鼓励,让我在刚刚进入到研究生阶段困惑迷茫的时候得以迅速适应和调整,获益良多。
最后,感谢我研究生阶段的朋友们,愿我们的友谊长存!





