
枯草芽孢杆菌(Bacillus subtilis)是革兰氏阳性细菌,是一种重要工业酶制剂的生产菌,中国农业部批准使用的安全菌株,现已广泛应用于食品发酵行业.
在基因工程操作中,因为高拷贝外源质粒在B. subtilis中的稳定性较差,造成复制过程中大量单链脱氧核糖核酸(deoxyribose nucleic acid,DNA)的产生。单链DNA的积累,最终导致质粒的不稳定,质粒的不稳定性成为基因工程操作的最大障碍.因此,对于B. subtilis的基因改造主要是针对染色体基因组的改造,将外源基因整合到B. subtilis菌株染色体上.
在前期的研究中,实验室已从贵州地区豆豉样品中分离获得可作为工业生产应用的菌株BJ3-2,但其谷氨酰胺酶活性较低。这就需要运用基因工程的方法对BJ3-2进行基因组改造,将高活性的glsA基因整合到BJ3-2中,形成新的工程菌株.考虑到BJ3-2应用于食品中的安全性问题,选择可通过升温消除的温敏型质粒载体pKSV7.研究采用同源重组技术有效改良食品级安全基因组,为下一步将高活性谷氨酰胺酶基因定点整合入BJ3-2菌株获得重组菌株提供了依据和理论基础。
1 材料与方法
1.1 材料与试剂
菌株:Escherichia coli DH5α、B. subtilis BJ3-2为本实验室保藏菌株。
质粒:pET28b为本实验室保存;pKSV7为军事医学科学院馈赠。
Taq聚合酶、限制性内切酶、DNA Marker:日本TaKaRa公司;基因组提取试剂盒:北京Promega生物技术有限公司;质粒提取试剂盒:美国OMEGA生物技术公司;氯霉素:北京SOLARBIO科技有限公司;细菌谷氨酰胺酶检测试剂盒:上海GENMED医药科技有限公司;其他试剂均为国产分析纯。
1.2 仪器与设备
MyCycler型PCR仪、Gel Doc XR型Universal Hood凝胶成像分析仪、165-2660型电穿孔系统(GenePulserXcell):
美国Bio-Rad公司;Sonifier 450D型超声破碎仪:美国BRANSON公司;SHZ-82型恒温水浴锅:江苏金坛市医疗仪器厂;DHP-9162型电热恒温培养箱:上海一恒科技有限公司;SKY-2102C型恒温摇床:上海苏坤实业有限公司;SW-CJ-IFD型标准型净化工作台:上海锦星科学仪器有限公司。
1.3 方法
1.3.1 目的基因组文库构建B. subtilis基因组DNA提取、质粒提取依照试剂盒说明书进行;DNA酶切、片段回收、连接采用常规方法。
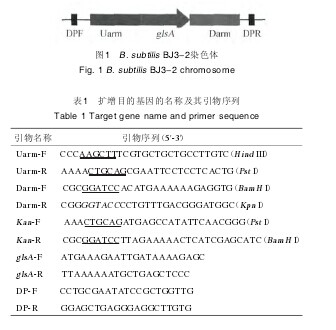
1.3.2 引物设计根据美国国家生物技术信息中心(national center ofbiotechnology information,NCBI)上查询得到的B. subtilis168菌株的基因组序列,定位glsA基因位置。截取glsA基因上游825bp片段作为上游同源臂Uarm;截取glsA基因下游902bp片段作为下游同源臂Darm;按pET28b载体上Kan基因序列设计引物;检测引物(detection primer,DP)为同源臂的上下游204bp和137bp位置处。基因构建的结构见图1,扩增基因名称和引物序列见表1.【图1.表1】
1.3.3 含Kan双交换整合载体(pKUKD)的构建以pKSV7质粒为骨架,分别克隆入Uarm、Darm片段和Kan基因3个基因片段。Kan为重组菌株BJ-Kan抗性筛选标记,构建过程见图2.【图2】
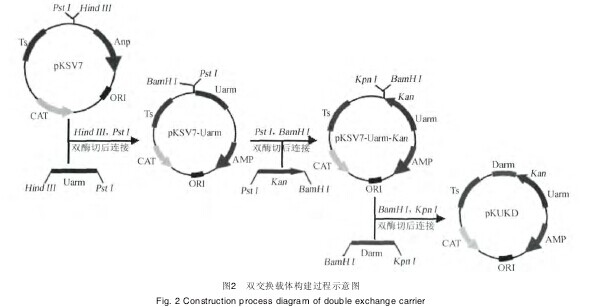
由图2可知,以B. subtilis BJ3-2基因组为模板,引物PUarmF/PUarmR和PDarmF/PDarmR扩增得到Uarm和Darm片段;以pET28b为模板,引物PkanF/PkanR扩增得到Kan基因.Hind III和Pst I对Uarm片段及pKSV7质粒进行双酶切、连接,并转化E. coli DH5α,获得pKSV7-Uarm重组质粒.Pst I和BamH I对Kan基因及pKSV7-Uarm质粒进行双酶切、连接,并转化E. coli DH5α。获得pKSV7-Uarm-Kan重组质粒.BamH I和Kpn I对Darm片段及pKSV7-Uarm-Kan质粒进行双酶切、连接,并转化E.coli DH5α。获得pKSV7-Uarm-Kan-Darm即pKUKD重组质粒。
1.3.4 测序及比对pKUKD质粒构建完成后,送至英潍捷基公司测序。测序结果通过BLAST程序与B. subtilis 168菌株进行同源性比对,验证构建序列是否正确.
1.3.5 同源重组介导glsA基因的敲除将pKUKD载体电转化到BJ3-2菌株感受态中.
菌体涂布于Luria-Bertan(iLB)固体平板上(含卡那霉素2μg/mL),通过双交换检测引物pDPF/pDPR筛选重组菌株。
当glsA基因被Kan基因置换后,通过聚合酶链式反应(poly-merase chainreaction,PCR)扩增得到约3000bp大小的片段;而当Uarm或Darm发生单交换时,PCR扩增得到12 000bp片段.
将验证后的阳性重组菌株45℃高温传代培养以丢失质粒,获得重组菌株BJ-Kan.
1.3.6 重组菌株BJ-Kan的稳定性检测将重组菌株BJ-Kan以0.1%的接种量接种至LB液体培养基中,连续传代20次后进行PCR检测。
1.3.7 重组菌株BJ-Kan及野生菌株BJ3-2的谷氨酰胺酶酶活测定将2种菌株在37℃、200r/min条件下振荡培养过夜,在波长600nm条件下测量其光密度值,OD600nm相同时依照BACTERIUM GLUTAMINASE ASSAY KIT说明书进行检测。
2 结果与分析
2.1 测序结果分析Uarm、Darm测序比对后发现,glsA基因上下游同源臂Uarm、Darm已正确连接,且与B. subtilis 168菌株同源性为99%,应为BJ3-2菌株上下游序列,可作为同源重组同源序列使用。
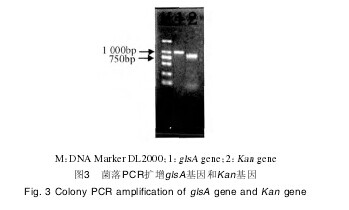
2.2 pKUKD载体转化B. subtilis BJ3-2转化子涂布氯霉素抗性平板,挑取单菌落,菌落PCR验证pKUKD载体是否成功转化至B. subtilis BJ3-2中。如图3所示,菌落PCR能扩增glsA基因及Kan基因,证明转化BJ3-2菌株中含有pKUKD质粒。【图3】
2.3 glsA基因敲除菌株的筛选与鉴定在所鉴定菌株中获得1株pDPF/pDPR引物扩增筛选的阳性菌株,菌落PCR扩增结果见图4.阳性菌株在3 000bp位置扩增出目的片段,且该菌株具有卡那霉素抗性.初步认定为glsA基因敲除菌株。将该阳性菌株接种于液体LB培养基,42℃高温过夜培养后,进行菌落PCR检测glsA基因及Kan基因。结果显示,Kan基因能有效扩增,而glsA基因无扩增,且BJ-Kan菌株已丧失质粒所带氯霉素抗性。证明该菌株为质粒已丢失的glsA敲除菌株。【图4】

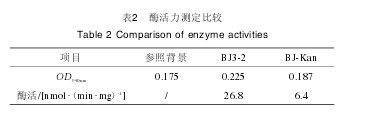
2.4 重组菌株BJ-Kan及野生菌株BJ3-2的谷氨酰胺酶酶活测定相同条件下的BJ-Kan菌株及BJ3-2菌株谷氨酰胺酶酶活力检测结果见表2.由表2可知,BJ3-2菌株及BJ-Kan菌株谷氨酰胺酶酶活力分别为26.8nmol(/min·mg)、6.4nmol(/min·mg),野生菌株BJ3-2酶活力较重组菌株BJ-Kan菌株高3.2倍,表明BJ-Kan菌株的glsA基因已被成功敲除。【表2】
3 结论
通过卡那霉素抗性筛选获得glsA基因敲除菌株BJ-Kan.证实了染色体上glsA基因可以通过所设计的上下游同源臂序列Uarm和Darm进行敲除、置换,为今后glsA基因的置换提供负筛选标记及理论基础.通过重组菌株BJ-Kan与原菌株BJ3-2的谷氨酰胺酶活力对比可以看出,原菌株的谷氨酰胺酶酶活较重组菌株高出3.2倍。因此,在忽略其他影响因素的情况下可以认为,B. subtilis的谷氨酰胺酶酶活受谷氨酰胺酶基因的调控,同源重组完全可以用来改变BJ3-2谷氨酰胺酶活性。
在对BJ3-2基因进一步改造的研究中,可利用得到的glsA敲除菌株BJ-Kan以及重新构建的双交换载体pKUGD(外源高活性谷氨酰胺酶基因载体),进行再次同源重组,从而获得含高活性谷氨酰胺酶基因的BJ-GA工程菌株。该菌株由于不存在外源质粒,且没有其他外源基因的插入(抗性基因),符合食品级改造的要求.因此,该菌株将可以用于食品(豆豉)发酵.
整合载体pKUKD的成功构建有利于外源高活性谷氨酰胺酶基因定点整合入B. subtilis染色体、在B. subtilis中稳定表达,为构建高表达效率的无抗性标记基因的工程菌奠定了重要的研究基础.但由于B. subtilis同源重组中双交换频率较低,pKUKD载体转化进入BJ3-2菌株后均可通过上游或下游同源臂与BJ3-2染色体形成单交换,从而导致双交换重组菌株的筛选还较为困难.
参考文献:
[1] HONG H A, HUANG J M, KHANEJA R, et al. The safety of Bacillussubtilis and Bacillus indicus as food probiotics[J]. J Appl Microbiol,2008, 105(2): 510-520.
[2] 董 晨,曹 娟,张 迹,等。 耐高温 α- 淀粉酶基因在枯草芽孢杆菌中的高效表达[J]. 应用与环境生物学报,2008,14(4):534-538.
[3] JOSEPH P, FANTINO J R, HERBAUD M L, et al. Rapid orientatedcloning in a shuttle vector allowing modulated gene expression in Bacil-lus subtilis[J]. FEMS Microbiol Lett, 2001, 205(1): 91-97.
[4] SOLAR G, ESPINOSA M. Plasmid copy number control an ever growingstory[J]. Mol Microbiol, 2000, 37(3): 492-500.
[5] YUE C Y, SUN M, YU Z N. Improved production of insecticidal proteinsin bacillus thuringiensis strains carrying an additional cry1c gene in itschromosome[J]. Biotechnol Bioeng, 2005, 92(1): 1-7.
[6] YUE C Y, SUN M, YU Z N. Broadening the insecticidal spectrum ofLepidoptera-specific Bacillus thuringiensis strains by chromosomal inte-gration of cry3A[J]. Biotechnol Bioeng, 2005, 91(3): 296-303.
[7] 贾东旭,吴拥军,李耀中,等。 细菌型豆豉发酵芽孢杆菌的筛选与鉴定[J]. 食品科学,2009,30(5):217-221.
[8] YAN X H, GAI Y L, LIANG L. A gene encoding alanine racemase is in-volved in spore germination in Bacillus thuringiensis[J]. Architect Mi-crobiol, 2007, 187(5): 371-378.
[9] 刘 萍,夏立秋,胡胜标,等。 外源基因在苏云金芽孢杆菌染色体上的定点整合及表达[J]. 微生物学报,2008,48(5):661-666.
[10] 沈关心,朱慧芬,张 悦,等。 菌落 PCR 和质粒 PCR 对转化菌的筛选[J]. 免疫学杂志,2000,16(2):149-151.
[11] 银 巍,黄奕俊,苏兴文,等。 T4 DNA 连接酶介导的 5'RACE 法克隆大鼠 Nor1 全长 cDNA[J]. 中国病理生理杂志,2004,20(3):317-321.
[12] XUE G P, JOHNSON J S, DALRYMPLE G P. High osmolarity im-proves the electro-transformation efficiency of the gram-positive bacte-ria Bacillus subtilis and Bacillus licheniformis[J]. J Microbiol Meth,1999, 34(3): 183-191.
[13] 卢 彪,吴拥军,刘艳敏,等。 高活性谷氨酰胺酶基因 glsA2在枯草芽孢杆菌 BJ3-2 染色体上的定点整合[J]. 食品科学,2013,35(1):141-144.
[14] PARK H W, GE B, BAUER L S, et al. Optimization of Cry3A yields inBacillus thuringiensis by use of sporulation-dependent promoters incombination with the STAB-SD mRNA sequence[J]. Appl EnvironMicrobiol, 1998, 64(10): 3932-3938.
[15] 李瑞芳,薛雯雯,黄 亮,等。 枯草芽孢杆菌感受态细胞的制备及质粒转化方法研究[J]. 生物技术通报,2011(5):227-230.





