
MicroRNA(miRNA)作为一种转录后调节因子在基因表达调控中发挥重要作用。它是一类广泛存在于真核生物中、长约22nt的内源性非编码小RNA分子,通过组装进RNA诱导的沉默复合体(RNA-in-ducedsilencingcomplex,RISC)与靶基因3'非翻译区(untranslatedregions,3'UTR)结合,根据miRNA与靶mRNA之间互补程度的不同导致靶基因降解或者阻遏靶mRNA的翻译.自1993年在线虫中首次发现miRNA至今,大量研究表明miRNA可通过精细调节基因的表达参与细胞发育、分化、增殖、凋亡以及应激反应等过程,其表达的组织特异性和时空特异性使其在多种生物学过程中发挥重要调节作用.据推测人类有超过1/3的基因受miRNA调控,而确定miRNA靶基因是研究miRNA生物学功能的关键。目前确定miRNA靶基因主要靠生物信息学预测和生物学实验方法进行验证,前者主要根据经实验观测得到的miRNA及其靶基因之间序列识别的规律性进行设计.应用较普遍的几个软件有miRanda,TargetScan和RNAHybrid.miRan-da作为第一个基于序列的预测算法从序列匹配,靶位点的保守性及双链结合的自由能角度进行分析,有比较好的检出率;TargetScan主要考虑物种间保守的miRNA靶基因,通过“种子匹配”可进一步增加预测精度;RNAHybrid的研究重点则在于RNA二级结构预测方面,通过动态规划寻找双链杂交的最优自由能鉴别miRNA的靶点,具有明显的速度优势.然而单纯依靠序列信息所得到的预测结果仍存在较多的假阳性,难以提高miRNA靶基因的预测效能。有研究表明,与基于序列的方法相比,利用在相同样本中检测到的miRNA和mRNA表达谱可以更准确地预测miRNA靶基因,更好地揭示miR-NA和靶基因的绑定关系.
管家基因(house-keepinggene)指的是在生物体几乎所有细胞中广泛而稳定表达的基因,也称为看家基因或持家基因,如微管蛋白基因、糖酵解酶系基因与核糖体蛋白基因等。这类基因对于维持细胞的基本结构和代谢,保证细胞存活生长并在维持细胞最低限度功能上发挥关键作用。目前国际上有关miRNA对管家基因调控的研究尚不充分,现有报道多集中于分别通过个别基因作为管家基因和非管家基因的代表来探讨miRNA的调控作用,如以编码核糖体蛋白的基因、氧化磷酸化的基因代表管家基因,以与神经元相关的基因代表非管家基因.尽管结果显示miRNA较少靶向编码核糖体蛋白的基因,但由于少数基因的代表性不强,因此这些研究的结论并不能很好地反映两类基因的整体特征。其次在Cheng等有关miRNA调控管家基因的研究中,虽然以整体管家基因作为研究对象,但仅单独利用软件进行预测所得到的miRNA对靶基因的调控关系中存在假阳性较高的问题,结果的可靠性值得怀疑,并且仅依赖miRNA绑定的数目作为评价指标难免导致结果存在偏差。
通常,生物在进化过程中需要适应各种多变的细胞环境,而在诸多组织中表达的基因在选择压力和个体适应性上表现出的显着优势使得它们能够在多种生存环境下发挥功能,保证机体正常运转。特别是在维持细胞基本结构和代谢功能中发挥保障作用的管家基因,有研究表明其表达的蛋白质同更多的分子发生相互作用并且在广泛改变的细胞环境下行使功能,这些基因受到各方面的约束限制从而减少了序列分歧.此外,miRNA和mRNA的3'UTR序列之间的识别机制主要在于miRNA核心区域即种子序列的匹配,生物进化过程中miRNA和靶基因所表现出的协同相互作用以及彼此的约束使得种子区域对突变非常敏感,稍有变异就会失去miRNA对其的调控,从而导致靶基因表达水平的剧烈变化.有鉴于此,从进化的层面进行研究有利于进一步揭示miRNA对管家基因调控的重要性。
考虑到已有研究中所存在的问题以及miRNA在基因表达调控中的重要作用,我们系统地整合了3个目前国际主流的基于序列的靶基因预测软件结果,并结合基因表达谱信息进行联合分析,同时还考虑到3'UTR长度的影响,将miRNA绑定的密度作为主要评定指标,从整体水平来研究miRNA对管家基因和非管家基因调控的差异,进一步利用数十个物种通过phastCons进化分析探讨3'UTR区域的保守性之间的差异,明确了这种调控差异与基因3'UTR进化保守性之间的关系。
1 材料和方法
1.1 数据来源和统计分析方法
参考Eisenberg等的工作共获得了人类575个管家基因的列表,该列表是目前国际上相关研究中最常用的管家基因列表,具体是通过使用Affy-metrixU95芯片的微阵列检测47个不同人类组织和细胞系的101个不同样本所产生的表达谱数据,以每个组织或细胞系的平均表达水平作为阈值,将在所有组织中都高于其相应平均水平的基因识别为管家基因。人类参考基因组数据来自NCBI下GRCh37版本(所对应的UCSC版本号:HG19),通过该数据库公布的信息对应得到566个管家基因并将剩余的32021个基因作为非管家基因,同时也分别获取了两类基因相应的3'UTR信息,经过EMBOSS软件去除polyA尾后得到3'UTR序列。miRNA靶的预测数据从miRNAMap2.0数据库下载得到,该网站存储了后生动物基因组中已通过实验验证的miRNA及其靶基因,并分别利用TargetScan、miRanda和RNAhybrid预测出miRBase9.2中公布的人类475个成熟miRNA序列对靶基因的调控关系。
mRNA和miRNA的表达谱数据分别来自NCBI的GEO数据库中GPL96平台下GDS596所提供的编码基因在79个人类组织中的表达水平以及使用Q-PCR的方法获得的240个miRNA在人类14个正常组织(包括心、肝脏、肾脏等)中的表达水平.数据统计分析的部分利用R语言实现,显着性检验采取wilcoxon秩和检验。
1.2 整合多种基于序列的预测方法识别miRNA靶基因
分别采用3个常用软件TargetScan、miRanda和RNAHybrid对靶基因进行预测,在识别靶位点时遵循以下原则:①每个靶基因上需包含多个靶位点;②通过Sfold实现靶位点的可接近性,即保证miR-NA所到达的区域是mRNA上易被绑定的区域。根据上述原则将得到的3个软件预测结果进行整合分析,根据“靶位点至少被3个软件中的两个预测到”这一条件对各软件预测结果取交集,从而在靶基因上筛选出相应的靶位点。考虑到同一miRNA会调控相同mRNA的不同位置,并且这些区域都可能被两个以上的软件预测到,我们把该区域的数目作为miRNA在靶基因上的绑定数量,最终将整合的靶基因预测结果同前述管家基因和非管家基因相对应,获得miRNA对这两类基因的调控关系。
1.3 基于序列和表达信息的联合分析预测miRNA靶基因
基因表达信息通过样本均一化处理,构建出各包含有14个组织的12625个mRNA和240个miR-NA的表达谱数据矩阵(miRNA:240×14;mRNA:12625×14),这些组织在两个表达谱矩阵中是一致的。采用Pearson相关系数构建mRNA和miRNA的相关性矩阵(12625×240),并根据以往报道中提到的在多数情况下miRNA对mRNA的负性调节作用,以0为阈值筛选出相关系数小于0的miRNA-mRNA作为潜在靶对。得到基于表达信息预测出的人类miRNA对靶基因的调控关系后,再同基于序列的预测结果取交集,重新获得miRNA在两类基因上的靶向信息。而后以-0.54作为相关系数阈值(通过样本相关性t检验得到,P=0.05)重复上述过程。其中在联合分析中,由于基于表达谱的预测没有显示miRNA在mRNA上绑定的位置信息,所以从miRNA出现的数目上进行统计,针对以上两种方法均呈现出的调控关系进行后续分析。
1.4 3'UTR区域的保守性分析
使用phastCons方法比较管家基因和非管家基因上3'UTR区域的保守性,这是基于phylo-HMM开发的用于描述基因组中每个位点下DNA替换的过程以及转变方式的概率模型,通过45个脊椎动物基因组的多序列比对描绘出每个位点下的保守性分值,反映物种系统发生过程;该方法不同于其他保守性打分的程序,不依赖于固定大小的滑动窗口,短序列高度保守的区域以及较长的比较保守区域可以获得较高的分值,可表征每个位点处碱基保守性的概率.从UCSC-Download下载人类基因组HG19版本下每条染色体包含的每个基因上各位点的phastCons分数,以0~1之间的数值表示,并将phastCons分数≥0.9的作为保守位点得到每条染色体上每个基因的保守区域信息;同时又从UCSC-TableBrowser中下载到相同参考基因组条件下每个基因3'UTR的位置信息。通过将以上两种信息应用到bedtools软件的coverageBed程序中并同管家基因和非管家基因相对应,最终分别得到两类基因的3'UTR上保守区域所占的比例,从而进行保守性比较。
2 结果
2.1 管家基因上miRNA的调控密度显着高于非管家基因
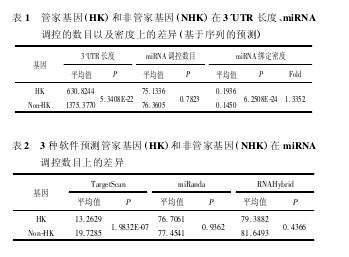
通过生物信息学软件预测寻找miRNA对两类基因的调控差异,对于475条人类成熟的miRNA序列,首先利用TargetScan、miRanda和RNAhybrid3个软件分别得到244389、1062973以及1009616个靶位点。为了降低假阳性,将各软件的预测结果取交集后分别得到miRNA对262个管家基因和7495个非管家基因的调控关系,结果发现两类基因中3'UTR的长度存在明显差异,与以往文献报道一致,同时发现两类基因在总体靶定的数目上并没有显着差异(表1).分别将3个软件各自的预测结果进行统计分析,发现彼此之间的预测结果并不完全一致(表2).为了排除3'UTR长度对总数目所产生的影响,利用miRNA调控的数目与UTR长度之比对miRNA在靶基因上的绑定情况做均一化处理,以miRNA绑定的密度作为衡量miRNA对两类基因调控差异的主要标准。结果发现管家基因上miRNA绑定的密度显着高于非管家基因(P=6.2508E-24,Fold=1.3352,见表1).
【表1 2】
2.2 整合基于表达谱的方法证明miRNA对两类基因调控的密度差异
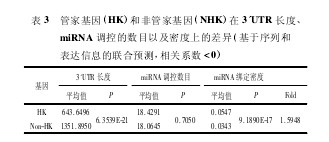
有研究表明加入表达信息能够有效提高预测精度,有利于更加明确miRNA和靶基因的绑定关系,为此在软件预测的基础上引入表达谱数据进行分析。由于miRNA负调控靶基因的表达,因此通过mRNA与miRNA的Pearson相关系数<0的指标在表达谱相关性矩阵中筛选两者之间的相互作用,分别得到miRNA对两类基因的调控(表3).这里所获得的候选样本数量同基于序列的预测结果基本持平,在wilcoxon秩和检验下254个管家基因的miRNA绑定密度也同样显着高于5688个非管家基因的,而数目分布依然没有差异,这与2.1的预测结果是一致的,并且倍率(Fold=1.5948)比之前有所提高,说明我们所采取的基于序列和表达谱信息的联合分析在预测miRNA对靶基因的调控上是可靠的。
【表3】
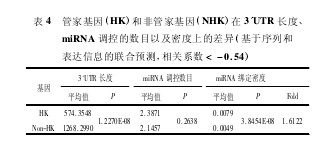
 为进一步优化预测结果,期望通过miRNA与mRNA之间具有统计显着性的负调节关系来反映生物细胞中更加真实的调控情况,利用14个组织的t检验得到的相关系数-0.54(P=0.05)作为双链之间反向调控的新临界值,构建出基于表达信息和软件预测的联合分析结果(表4),结果可见93个管家基因和1819个非管家基因在miRNA绑定密度的统计学检验中依然呈现出与之前预测结果相同的趋势。样本数量的下降使得P值(P=3.8454E-08)虽然没有之前显着(P=9.1890E-17),但是Fold值得到了进一步提高(Fold=1.6122).
为进一步优化预测结果,期望通过miRNA与mRNA之间具有统计显着性的负调节关系来反映生物细胞中更加真实的调控情况,利用14个组织的t检验得到的相关系数-0.54(P=0.05)作为双链之间反向调控的新临界值,构建出基于表达信息和软件预测的联合分析结果(表4),结果可见93个管家基因和1819个非管家基因在miRNA绑定密度的统计学检验中依然呈现出与之前预测结果相同的趋势。样本数量的下降使得P值(P=3.8454E-08)虽然没有之前显着(P=9.1890E-17),但是Fold值得到了进一步提高(Fold=1.6122).
【表4】
2.3 管家基因3'UTR上的保守区域显着高于非管家基因
为了从进化层面探索miRNA对管家基因的调控,进一步考察miRNA对两类基因调控的密度差异同3'UTR区域的保守性之间的关系,利用phastCon分数比较两类基因3'UTR区域的保守性,结果可见3'UTR上的保守区域分布中566个管家基因的保守区域(phastCon分数≥0.9)平均占有比例为25.60%,显着高于32021个非管家基因(保守区域平均占有比例=16.75%,P=5.1658E-26).同时,对保守位点的统计发现在两类基因的3'UTR上只存在非保守区域(phastCon分数<0.9)的基因分别占了8.5%(HK)和19.5%(Non-HK),从而反映出管家基因中保守性序列占有的区域较大,3'UTR在进化中也就更加趋于保守。由于3'UTR是miRNA与靶基因相互作用的主要区域,是miRNA参与基因调控的主要调控元件,因此其在管家基因中的进化保守性较高提示可能与管家基因miRNA调控密度具有相关性。
3 讨论
管家基因作为维持细胞最低限度功能并使其存活生长所必需的蛋白质编码基因,其表达产物对于生物体的基本生命活动是必须的,而作为基因转录后调控因子的miRNA通过精细调节基因表达在诸如生长发育等多种生物学过程中发挥着重要作用。
鉴于每个miRNA可以调控几十个甚至上百个靶基因,因此准确鉴别出miRNA在管家基因和非管家基因上的靶点具有重要意义。之前有关此方面的报道有的专注于个别基因进行讨论从而不具有代表性,有的只采取单一的靶基因预测软件导致miRNA对管家基因的调控结果不可靠,并且单纯以miRNA绑定的数目作为衡量指标而没有考虑3'UTR的长度来进行研究,降低了结果的可信度。本研究在考虑到上述问题的基础上从全局的整体水平来分析miRNA对两类基因的靶向性特点,理论上更具有代表性。
通过生物信息学计算预测的方法分析miRNA对管家基因和非管家基因的调控,结果发现不论单纯基于多个软件预测还是整合表达信息的方法都显示出miRNA在管家基因和非管家基因上的调控存在明显不同,具体表现为管家基因3'UTR区域miR-NA绑定的密度显着高于非管家基因。在基于序列的靶位点识别中,分别将TargetScan、miRanda和RNAhybrid3个软件各自的预测结果进行统计分析,发现不仅彼此之间的结果并不完全一致,而且各软件的单独预测结果也都同之前文献报道中所采用PITA进行预测的结果不一致(Cheng等的工作发现miRNA对管家基因调控的数目显着低于非管家基因,密度上没有表现出明显差异),更加说明应用单一软件识别靶位点的方法是不准确的,结果的可信性值得怀疑。本文整合多种软件进行分析并引入表达信息提高了预测精度,Fold值的不断改善也表明了该方法使得靶基因的预测结果趋于可靠,当然也有可能遗漏掉部分真正的miRNA靶基因。
由于管家基因和非管家基因在3'UTR长度上有明显差异,考虑到长度不同对起调控作用的miRNA数目的影响,研究发现以单位位点上miRNA的调控数目作为评定指标更加合理。Stark等尽管也通过miRNA绑定密度来研究调控差异,但结果却与我们的相反,主要是由于选取的背景基因不同,他们重点关注了部分基因,利用GO分支中属于编码核糖体蛋白的128个基因和神经元相关的364个基因来比较差异,并根据编码核糖体蛋白的基因上miRNA绑定的密度显着低于后者的结果来反映miRNA对管家基因和非管家基因的调控差异,容易导致以偏概全。我们则是将所有管家基因作为分析对象,避免以局部的结果来反映整体的调控水平。
考虑到miRNA的长度限制,将比较表达谱数据引入前后的预测结果可以看出miRNA的绑定密度指标得到改善,例如表1中HKmiRNA绑定密度为0.1936,在表3中降低到0.0079,这种结果更加符合miRNA在mRNA上的实际靶向情况。此外由于miRNA在动物中常结合于3'UTR区域,尽管管家基因的3'UTR在polyA尾去除前后均较短,理论上相对于非管家基因应该会逃逸miRNA的调控,之前的报道都倾向于这种结论,但我们的预测从另一个侧面显示了miRNA的调控对于管家基因的重要作用。同时我们还发现两类基因在miRNA靶定密度上的差异与3'UTR的序列保守性具有一定的相关性,这种相关性的出现可能是源于生物不断进化过程中管家基因需应对各种环境的改变,在多种约束条件下各组织中广泛表达的基因要行使基本功能使得序列之间的分歧度减少,碱基更不容易发生变异,尤其是miRNA在对管家基因转录后水平的调节中双链之间核心区域的匹配非常重要,若出现突变将失去这种调控作用,在协同进化过程中不能随意突变保证了一部分miRNA对mRNA的调控。在3'UTR区域的进化分析中也看出管家基因中保守区域所占的比例较多,可见miRNA对管家基因的调控很必要,并且与3'UTR区域的保守性是相关的。更重要的是近几年有关miRNA对基因表达调控的理论研究表明,miRNA对靶基因的负性调控作用同转录因子的转录调控相联合,形成了具有特定拓扑结构的网络模体,在增强靶基因表达稳健性的方面发挥鲁棒性调控作用.综上所述,我们的研究结果进一步突出了miRNA在管家基因调控中的重要性,提示其可能在维持管家基因表达的稳定性上起重要作用。
本研究反映出3'UTR区域的保守性与管家基因中miRNA调控密度较高具有相关性,但通过多种软件预测所得到的结果以及表达谱的数据并不能直接表明两者之间的因果关系,这一问题的解决还需要后续工作从基因进化和miRNA-靶基因相互作用的分子机制方面做进一步证明。同时由于真核基因表达调控的复杂性,仅从miRNA角度研究靶基因的表达调控作用是不完全的,还需联合分析转录因子对表达水平的影响以及在维持基因表达的动态平衡中所发挥的调节作用,从而进一步揭示管家基因的调控机制。
【参考文献】
[1]BaggaS,BrachtJ,HunterS,etal.Regulationbylet-7andlin-4miRNAsresultsintargetmRNAdegradation[J].Cell,2005,122(4):553-563.
[2]LaiEC.MicroRNAsarecomplementaryto3'UTRsequencemo-tifsthatmediatenegativepost-transcriptionalregulation[J].NatGenet,2002,30(4):363-364.
[3]AmbrosV.ThefunctionsofanimalmicroRNAs[J].Nature,2004,431(7006):350-355.
[4]WienholdsE,PlasterkRH.MicroRNAfunctioninanimaldevel-opment[J].FEBSLett,2005,579(26):5911-5922.
[5]夏伟,曹国军,邵宁生。MicroRNA靶基因的寻找及鉴定方法研究进展[J].中国科学(C辑:生命科学),2009(1):121-128.
[6]MendesND,FreitasAT,SagotMF.Currenttoolsfortheidentifi-cationofmiRNAgenesandtheirtargets[J].NucleicAcidsRes,2009,37(8):2419-2433.
[7]PengX,LiY,WaltersKA,etal.ComputationalidentificationofhepatitisCvirusassociatedmicroRNA-mRNAregulatorymod-ulesinhumanlivers[J].BMCGenomics,2009,10(1):373.
[8]SmeetsA,DaemenA,VandenBemptI,etal.Predictionoflymphnodeinvolvementinbreastcancerfromprimarytumortis-sueusinggeneexpressionprofilingandmiRNAs[J].BreastCancerResTreat,2011,129(3):767-776.
[9]StarkA,BrenneckeJ,BushatiN,etal.AnimalMicroRNAsconferrobustnesstogeneexpressionandhaveasignificantimpacton3'UTRevolution[J].Cell,2005,123(6):1133-1146.
[10]ChengC,BhardwajN,GersteinM.TherelationshipbetweentheevolutionofmicroRNAtargetsandthelengthoftheirUTRs[J].BMCGenomics,2009,10:431.
[11]KumaK,IwabeN,MiyataT.Functionalconstraintsagainstvari-ationsonmoleculesfromthetissuelevel:slowlyevolvingbrain-specificgenesdemonstratedbyproteinkinaseandimmunoglobu-linsupergenefamilies[J].MolBiolEvol,1995,12(1):123-130.
[12]HastingsKE.StrongevolutionaryconservationofbroadlyexpressedproteinisoformsinthetroponinIgenefamilyandothervertebrategenefamilies[J].JMolEvol,1996,42(6):631-640.
[13]DuretL,MouchiroudD.Determinantsofsubstitutionratesinmammaliangenes:expressionpatternaffectsselectionintensitybutnotmutationrate[J].MolBiolEvol,2000,17(1):68-74.
[14]HughesAE,BradleyDT,CampbellM,etal.MutationalteringthemiR-184seedregioncausesfamilialkeratoconuswithcataract[J].AmJHumGenet,2011,89(5):628-633.
[15]LewisMA,QuintE,GlazierAM,etal.AnENU-inducedmuta-tionofmiR-96associatedwithprogressivehearinglossinmice[J].NatGenet,2009,41(5):614-618.
[16]MenciaA,Modamio-HoybjorS,RedshawN,etal.MutationsintheseedregionofhumanmiR-96areresponsiblefornonsyndromicprogressivehearingloss[J].NatGenet,2009,41(5):609-613.
[17]EisenbergE,LevanonEY.Humanhousekeepinggenesarecom-pact[J].TrendsGenet,2003,19(7):362-365.
[18]SuAI,CookeMP,ChingKA,etal.Large-scaleanalysisofthehumanandmousetranscriptomes[J].ProcNatlAcadSciUSA,2002,99(7):4465-4470.
[19]WodickaL,DongH,MittmannM,etal.Genome-wideexpres-sionmonitoringinSaccharomycescerevisiae[J].NatBiotechnol,1997,15(13):1359-1367.
[20]LockhartDJ,DongH,ByrneMC,etal.Expressionmonitoringbyhybridizationtohigh-densityoligonucleotidearrays[J].NatBiotechnol,1996,14(13):1675-1680.
[21]OlsonSA.EMBOSSopensupsequenceanalysis.EuropeanMo-lecularBiologyOpenSoftwareSuite[J].BriefBioinform,2002,3(1):87-91.
[22]HsuSD,ChuCH,TsouAP,etal.miRNAMap2.0:genomicmapsofmicroRNAsinmetazoangenomes[J].NucleicAcidsRes,2008,36(Databaseissue):D165-D169.
[23]LewisBP,ShihIH,Jones-RhoadesMW,etal.PredictionofmammalianmicroRNAtargets[J].Cell,2003,115(7):787-798.
[24]JohnB,EnrightAJ,AravinA,etal.HumanMicroRNAtargets[J].PLoSBiol,2004,2(11):e363.
[25]KrugerJ,RehmsmeierM.RNAhybrid:microRNAtargetpredic-tioneasy,fastandflexible[J].NucleicAcidsRes,2006,34(WebServerissue):W451-W454.
[26]Griffiths-JonesS.miRBase:themicroRNAsequencedatabase[J].MethodsMolBiol,2006,342:129-138.
[27]BarrettT,EdgarR.Geneexpressionomnibus:microarraydatastorage,submission,retrieval,andanalysis[J].MethodsEnzy-mol,2006,411:352-369.
[28]SuAI,WiltshireT,BatalovS,etal.Ageneatlasofthemouseandhumanprotein-encodingtranscriptomes[J].ProcNatlAcadSciUSA,2004,101(16):6062-6067.
[29]LuJ,GetzG,MiskaEA,etal.MicroRNAexpressionprofilesclassifyhumancancers[J].Nature,2005,435(7043):834-838.
[30]DingY,LawrenceCE.AstatisticalsamplingalgorithmforRNAsecondarystructureprediction[J].NucleicAcidsRes,2003,31(24):7280-7301.
[31]MuniateguiA,PeyJ,PlanesFJ,etal.JointanalysisofmiRNAandmRNAexpressiondata[J].BriefBioinform,2013,14(3):263-278.
[32]BartelDP.MicroRNAs:genomics,biogenesis,mechanism,andfunction[J].Cell,2004,116(2):281-297.
[33]KimVN,NamJW.GenomicsofmicroRNA[J].TrendsGenet,2006,22(3):165-173.
[34]SiepelA,BejeranoG,PedersenJS,etal.Evolutionarilycon-servedelementsinvertebrate,insect,worm,andyeastgenomes[J].GenomeRes,2005,15(8):1034-1050.
[35]BlanchetteM,KentWJ,RiemerC,etal.Aligningmultiplege-nomicsequenceswiththethreadedblocksetaligner[J].GenomeRes,2004,14(4):708-715.
[36]HuangJC,BabakT,CorsonTW,etal.Usingexpressionprofi-lingdatatoidentifyhumanmicroRNAtargets[J].NatMethods,2007,4(12):1045-1049.
[37]BerezikovE.EvolutionofmicroRNAdiversityandregulationinanimals[J].NatRevGenet,2011,12(12):846-860.
[38]KloostermanWP,PlasterkRH.ThediversefunctionsofmicroR-NAsinanimaldevelopmentanddisease[J].DevCell,2006,11(4):441-450.
[39]ZhangL,HammellM,KudlowBA,etal.SystematicanalysisofdynamicmiRNA-targetinteractionsduringC.elegansdevelop-ment[J].Development,2009,136(18):3043-3055.
[40]EbertMS,SharpPA.RolesformicroRNAsinconferringrobust-nesstobiologicalprocesses[J].Cell,2012,149(3):515-524.
[41]JungM,SchaeferA,SteinerI,etal.RobustmicroRNAstabilityindegradedRNApreparationsfromhumantissueandcellsam-ples[J].ClinChem,2010,56(6):998-1006.





