����ժ Ҫ����������֬�ʴ�л����Ҫ����������֬�ʴ�л���һᵼ��֬���Ρ��ι���˥�ߵ����ؼ�����������ά�ָ���֬�ʴ�л��̬�з�����Ҫ���ã�������ǿ�����ƿ�Ӱ��֬�ʴ�л���������;���в���ȫ�������ʳ��������ҩ������ؿ���ͨ��AMPK��m TOR��PPARα��SIRT1��������Ӧ��������;��Ӱ�����֬�ʴ�л��֬�ʴ�л���Ҷ�����Ҳ��Ӱ�졣���ľͽ���������Ӱ��֬�ʴ�л;�����о���չ��һ������
�����ؼ��ʣ�������; ����; ֬�ʴ�л;
��������������֬�ʴ�л���������٣���֬������������ա����估�ֽ�ȴ�л����������Ҫ���ã�����֬�ʴ�л���һ�����֬���Ρ���Ӳ�������ؼ��������꣬�о��߷���������ά�ָ���֬�ʴ�л��̬���к���Ҫ�����á���ʳ��������ҩ������ؿ�ͨ��Ӱ�����ɣ����ڸ���֬�ʴ�л�����ľͽ�������Ӱ��֬�ʴ�л;�����о���չ��һ������
����һ�����������ص�
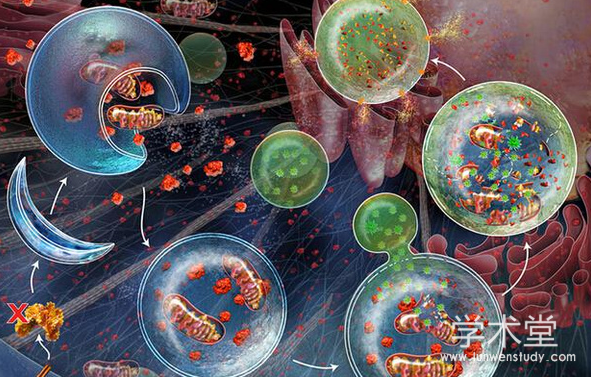
����������һ�ָ߶ȵ��ڡ����ص�ϸ�����̣���ͨ����ø�彵��ϸ������֣�ά��ϸ������������ƽ�⡣���ɿ�Ӱ����Ҫϸ�����̣�������֢������Ӧ������������֬�ʴ�л�ʹ洢����������̬���ۼ���������ȡ�Singh��(2009)�����Ӫ��ȱ��ʱ��ϸ����֬�����Ը�������(triglyceride,TG)��ʽ�����֬��ˮ���֬�����Ի������������ʱ��������������ϸ���ڵ����ʺ�ϸ����������˫Ĥ�����У�������ø���Խ���ܡ����ɿ�ѡ�����������ض�������磬���Ӱ��½鵼������(chaperone-mediated autophagy,CMA)��һ��ѡ�������ɣ�ֻ�е�������ø�彵�⡣�����зḻ����ø���ˮƽ�����ɡ�������ά�ָ���֬�ʴ�л����̬�з�������Ҫ�����ã���ǿ���ɿ����Ӹ���֬�����ģ����ٸ���֬�ʻ��ۣ��������ɻ��������֬�ʻ��ۣ�����ʱ�ᵼ�¼�����
������������֬�ʴ�л
��������������֬�ʴ�л���������٣���֬��ĸ��ִ�л����������Ҫ���á������зḻ��øϵͳ���Ǻϳ�֬���ᡢ���̴�����֬����֭������֬���̴�֬����ת��ø(lecithin-cholesterol acyltransferase,LCAT)����Ҫ������������ͪ�塢�����ֽ�֬�������Ҫ���١�Schneider��(2014)���֬�ʴ�л�Ĺؼ�øͨ����CMA���⣬��CMAȱ�ݶ����пɹ۲쵽���֬���л�쳣����Щ����ָʾCMA�������֬�ʴ�л�ĵ��ڡ�֬��������һ�����������֬��(lipid droplet,LD)�����ɣ����·��ֵ�ϸ��֬�ʴ�л�����ߡ��ڸ��࣬֬�����ɺ�֬��ø������֬���ֽ�ͬ����֬�ʴ�л�з�����Ҫ���á�
��������Ӱ��֬�ʴ�л������;��
����Ӱ�����ɵ�������Ҫ�������࣬һ��ҩ��Ӱ�죬������ʳ������Ӱ�졣��Щ���ؾ��ɵ������ɻ��ԣ�Ӱ�����֬�ʴ�л��̬�����ڵ�;����:(1)AMPK;��;(2)m TOR;��;(3)PPARα;��;(4)SIRT1;��;(5)������Ӧ��;(6)����;����
����(һ)AMPK;��
��������-���ἤ��ĵ���ø(adenosine 5'-monophosphate(AMP)-activated protein kinase,AMPK)�����ڵ���������л����Ҫ���ӡ�AMPK�ܱ����ڸ������ʼ����ͨ��������ص���1(Atg1)���������ɵļ�ø��
����Wei��[1]����Zn[2]+��֬�����ɵ�ǿ�����ӣ�ͨ������Ca[2]+/CaMKKβ/AMPKͨ·���յ�֬�����ɡ�Huang��[2]��С���ϸ�����о����������4-�ȶ���़��Ḵ����(4-chlorodipicolinic acid,VOdipic-Cl)ͨ������LKB1/AMPK�������ź�ͨ·�����ɣ�������֬�ʻ��ۡ�Lee��[3]���ֶ�ʮ���鴼������AMPK-β�ǻ��ı���λ�㣬����AMPK��ͨ�����ữ����3-�ǻ�-3-���������øA��ԭø�Ļ��ԣ��յ��������ɣ����ٸ���֬�ʻ��ۡ�Chen��[4]����5-��������-4-������-1-4-ૺ�����(5-aminoimidazole-4-carboxamide ribonucleotide,AICAR)����AMPK����������ά�����������LX-2ϸ����ת����������β(transforming growth factor-β��TGF-β)������������ġ�Li��[5]���ֶ���˫�ҿ���ǿ�������ɡ�����֬����֯���ɣ�ʹ����������ص���1����3(LC3)��AMPK�ı���ˮƽ�������ߣ��Ӷ����Ƹ�֬��ʳ(high fat diet,HFD)�յ��ĸ���֬�����Ժ�Ѫ��TGˮƽ��
����WD40-�ظ��������漰����ϸ����Ĵ��͡��߶ȱ��ص��ν��Ӽ��壬RACK1�ǽ���WD40�ظ����еĵ����ʡ�Zhao��(2015)����RACK1����ǿAMPK���ữ���Atg14L-Beclin 1-Vps34-Vps15�������γɣ��ٽ����ɡ�RACK1��Thr50����AMPK���ữ��������������︴������γɡ�RACK1��Thr50���ữ��ǿ����Vps15,Atg14L��Beclin 1ֱ�ӽ�ϣ��ٽ�������ʼ���������װ��
�������⣬�����仨�۶��ǡ�θ�����ء�TGF-β���ø-1(TAK1)�ȣ����ܼ���AMPK��
����(��)m TOR;��
�������鶯������ù�ذб�(mammalian target of rapamycin,m TOR)��������m TOR������룬��һ�ֵ���ø��m TOR�����������ʽ�ϣ��γ�m TOR������1��m TOR������2��m TOR��Ϊ���ָ�����ĺ��ģ�����˿����/�հ��ᵰ��ø���ã����ڸ�ϸ�����ɡ���AMPK;����ͬ������m TOR�������ɣ�����m TOR�������ɡ�����ù�ؿ���m TOR������е��ϣ���m TOR���Ƽ���
����Rujano��[6]�о���������ʧ������ø���ữȱ����m TOR�źŴ����йء�ATP6AP2����ͻ��ʹV-ATPø��װ�����������ǻ���������ȱ�ݡ�Wang��(2016)���֣�CYP7A1�������ż�����������/AKT�źŶ�m TOR���ưб�ļ��Wangȷ����һ���µ�CYP7A1-AKT-mTOR�ź��ᣬ���ź����ѡ�����յ��������ɡ�AICAR���Ƽ�������ù��/Aktͨ·�Ļ�еĿ�꣬����TGF-β�յ�������ͨ������ø����֬�ν���[4]��
����Zhong��[7]���ֶ�ɳ̹(Irb)ͨ������PPAR-y��p-AMPK������p-Akt��p-mTOR��ʹLC3BIl��LC3BIl/l��ֵ���ӣ����Է���2������db/dbС��������յ����ɡ�Irbͨ���ϵ�PPAR-y�������AMPK/Akt/m TOR�ź�ͨ·���յ��������ɡ�He��(2016)��ʵ����ʾ������ͨ������ERK/m TORͨ·���յ���ˮƽ������ͨ��������������ɸ��Ƹ���֬�ʻ��ۡ�Liu��(2016)������ά�����آ��ͼ�����5(FNDC5)ȱ��֢��ͨ��m TOR;���������ɡ�Song��(2015)���ֶ���˫�ҿɼ���PRKA��ͨ��PRKA-mTOR-ULK1�źŴ���;���յ����ɡ�
����ͬʱ��ľͨ����D���ȸ�Ѫ��������-1�������������m TOR�ź�ת����
�������⣬����m TOR���µ�V-ATPø����ƻ���ø��������ữ���������ɹ����ϰ�����������m TOR����ǿ���ɣ����ٸ���֬�ʻ��ۡ�InokuchiShimizu��(2014)���ָ�ϸ��������ȱʧTak1�Ľ�ʳС����ֳ����صĸ�ϸ������֢������WT��������ȣ�����m TORC1�������Ӻ��������ơ��ɼ�������AMPK��m TOR�ڵ��������������෴�����ã�AMPK��m TOR�Э����ͬ�������ɻ��ԡ�
����(��)PPARα;��
����PPARα(peroxisome proliferator-activated receptorα)����������ø����ֳ����������α����һ�������յ��ĺ����嵰�ף���PPAR��һ�����͡�PPARα����������ɵ���———����������ص����ʵĻ���ת¼������֬����β-�����������������ɡ�PPARα����Ϊ�����ź������ɻ���ת¼֮���Ŧ����
����Wei��[1]����п��ͨ������֬������Zn[2]+/MTF-1/PPARα;�����ٸ���֬�ʳ�����Zn[2]+ͨ����ǿ������ӦԪ�����ת¼����MTF-1��PPARαDNA����������Ľ�ϣ��������ɺ�֬���ֽ�Ĺؼ�����ת¼��Hsiao��[8]��ʵ����ʾ������ͪͨ��PPARα��PPARγ�����ķ�ʽ��ǿϸ����֬�ʷֽ⡢β-���������ɣ��������֬�����ԡ�Cho��(2017)��ʵ��֤��PPARα��SIRT1�źŴ���Ҳ�е������á�
�����о���Ҳ��������PPAR����ǡ���෴�ĺ�����———���ᴼX����(FXR)��Lee��(2014)����PPARα��FXR�ֱ��ڽ�ʳ��ι���ĸ����б����PPARα��FXR������С��������ɣ�ǰ�ߵ�ҩ��ѧ���ת�˽�ʳ״̬�����ɵ��������ƣ��յ�����֬�ʽ������֬�������ߵ�ҩ��ѧ�ǿ�������ڽ�ʳ״̬�µ������յ���PPARα��FXR����������ɻ����������еĹ���λ�㣬�����෴��ת¼�������Щ�����ʾ��ͨ��Ӫ��״̬�������ɵĻ������������ơ�
����(��)SIRT1;��
����SIRT1(sirtuin type1)�dz�Ĭ��Ϣ��������-2����ij�Ա�����������������ʶ�������(NAD+)�������鵰��/��������������ø�������ʺ��鵰��ȥ������ʹ����ת¼�͵����ʹ����ϵ����µ���Colak��(2014)����SIRT1��ȥ�������������ÿ��յ��������ɣ����ٸ���֬�ʻ��ۡ�Sun��[9]����SIRT1���ڻ������ڽ�ʳ��Ӧ����ǿ���ɺ����Ƹ���֬�ʴ�������DZ���ġ������شٽ�SIRT1ȥ����������ͨ��������ص���5(Atg5)�����Է�ʽ�յ������Ե���֬�����á�Ding��[10]��ʵ���У���«��(һ����Ȼ���)���������Ƽ���SIRT1���յ����ɣ���֤����������SIRT1-FoxO�źŴ���;���յ��ġ�SIRT1����;����Ҫ�е���������(30%)�Ͱ�«��(ҩ��ѧSIRT1�����)�ı������á�����ѧ�߷��ֶ���˫��Ҳ��ͨ��SIRT1-FoxO�źŴ���;���յ����ɡ�Sathyanarayan��[11]����֬����������֬ø(adipose triglyceride lipase,ATGL)��ǿSIRT1���ԣ�֤��SIRT1��ATGL�鵼�����ɺ�֬�������DZ���ģ�ͬʱATGLͨ��SIRT1�ٽ������Կ��Ƹ���֬�ηֽ��л��Zhang��(2015)������Ȼ��Ӱ�«��(resveratrol,RSV)������cAMP��SIRT1�����ữ����øA(pPRKA)�����ữAMP�����ø(pAMPK)ˮƽ���Լ�HepG2ϸ����SIRT1���ԣ���ͨ��cAMP-PRKA-AMPK-SIRT1�ź�ͨ·�յ����ɡ�
�������⣬�˲�����Rb2��2-�������ƶ����ȣ����ܼ���SIRT1��
����(��)������Ӧ��;��
����������(ER)��һ�ֶ�̬ϸ��������֬�ʴ�л��ԭʼλ�ã�֬�ʴ�л���ø�����ڵ��ֲ������ϡ�������Ӧ��(endoplasmic reticulum stress,ERS)���ձ���������ϸ���е�Ӧ��-�������ơ�Դ��ER�������������۵������Ŷ����յ���֢������Ӧ��[12]����ѹ������£�ER�������𣬵����ʳ������𣬵��´����۵��ĵ����ʻ��ۣ�����������Ӧ����Ӧ��Ϊδ�۵������ʷ�Ӧ(UPR)���о����֣�ERSʱϸ��ͨ���ϵ�GRP78�ȷ��Ӱ��µı������ٽ�δ�۵�/�����۵���������ȷ�۵�����GRP78�����ϵ�����Ϊ������Ӧ�������ı�־��ͬʱ����ͨ��ϸ���ڵĵ����ʽ���ϵͳ��δ�۵�/�����۵������н��⣬��������������[13]�����ڻ�����̬��ƻ�ʱ��δ�۵������ʷ�Ӧ��3���ź�ͨ·������ת¼����6(ATF6)������øRNA��ER��ø(PERK)�ͼ�����Ҫø-1α(IRE1α)���ֱ�ͨ����ǿ�������۵����������ٵ��������ɺʹٽ���������ص����ʽ����;������ϸ����ѹ�����ɵij̶Ⱥ�ϸ����ѹ��ˮƽ�й�[12],UPR����ϸ������ѹ����������ϸ����̬�ؽ���Ȼ�����ڳ���ERS�ڼ䣬UPR��ִٽ�ϸ��������ERS�ɵ������ɣ��յ�ϸ������������������Ӧ��ͨ��δ�۵������ʷ�Ӧ�Ͱ�����Ca[2]+Ũ�ȱ仯������ط����źŵ������ɣ��ٽ�������ص����ʱ���յ����������ɿ���ͨ�����������ڻ��ƣ�����������Ӧ����ص����ʱ���������Ӧ��ˮƽ�Ӿ磬ά��ϸ�����ܣ��ֿ�ϸ���������������漰�����������źŻ����һ���о�[12]��Kwanten��(2016)��ΪERӦ���յ������Իָ�ϸ�������ԣ�ERӦ���ȿ��յ�Ҳ���������ɣ���ʹ����ѡ���Եķ�ʽ�������Ҳ���ڣ���������Ӱ��ER��ת���ʹ����۵�����ȥ��������ERS��
����C1q/TNF��ص���9(C1q/TNF-Related Protein 9,CTRP9)��Ԥ���Ǿƾ���֬���Ըβ�(nonalcoholic fatty liver disease,NAFLD)��Jung(2015)����HFDι��С��ĸ����У��ٲ����鵼��CTRP9�����������յ�AMPK���ữ�����ɣ�����ERS��ǡ�����CTRP9ͨ��AMPK�鵼�������յ�����ERS���������֬�����ԡ�
����(��)����;��
��������ø����(Pip4k2a��Pip4k2b)ȱʧ��ʹ���������ȱ�ݣ�����ֹͣ�����½�ʳ�ڼ�С�������֬�κ�����С�ݴ�������[14]���Ҵ�����Rab7���ԣ�ʹ��������LDs�����䡢������ں�������������[15]��p300������VPS34������/ȥ��������VPS34�������ѧ�ؼ����������ž������ɺͷǾ������ɵ�������p300��ʧ���յ�VPS34������������������[16]��Lin��(2016)��ΪGTPase����M(IRGM)��������Ӱ�����ɵ��ڣ���������Ǿƾ���֬���εķ��ա�Martinez-Lopez��(2016)����������ϵͳ(CNS)�е��ض���Ԫ�ɶԸ����е�֬�����ɽ���ң����ơ���ø�����Ĥ����3(LAMP3)�������HepG2ϸ���м���Akt���ϵ�֬������øFASN��SCD-1�ı��ͨ������PI3K/Akt;�����ڸ���֬�ʴ�л[17]��Ca[2]+��Ca[2]+��ϵ��ɵ��������ź�ͨ·��������������������ø������ںϺ��������á�������·���У���ѧ���ڸ�ϸ��������������з�����AnxA6��AnxA6������Ĥ�������ܴ������µ�Ca[2]+ЧӦ���ӣ��������ɺ�ϸ�����̹��̵���������[18]���ڶ��ش���(5 mg/kg)�鵼��TLR4�̼����յ����ɣ��������ڰ�Ѫ֢�ڼ�ά��֬�ʴ�л��̬[19]��PLIN2�����ﱣ��֬��С�����ܾ�����/���ɣ�PLIN2ȱ������ǿ���ɲ����ĸ���TG����plin2-/-С������ǿ���ɿ�Ԥ�����ص�ERӦ���յ��ĸ�ϸ������֢��ϸ������[20]��������֬����ͱ���֬�������������ɺ�ϸ��������Mei��(2011)���ֲ�����֬����———���ᣬ�ٽ�����TG��֬���γɲ��յ�����;����֬����———����ᣬ����ת��Ϊ����TG��֬�Σ��������ɲ������յ�ϸ��������Tan��(2012)��������ͨ������øC�鵼���źŴ���;���յ����ɡ�Shen��(2016)����������ʹHsp27���ữ���ٽ����յ������ɡ���Ӱ�����ɵ����ػ��кܶ࣬������ͨ�����ͼ���֬���ء���֬������-5-����4-��ø����״�ټ��ء���(IV)-�ȶ��������εȡ����⣬������ι�������������֬����(PUFA)����ʳ���˶���������С�����Ⱦ���ֱ�ӻ��ӵ������ɡ�
�����ġ����ɶ�֬�ʴ�л��Ӱ��
����(һ)����֬�ʵ�����ϳ�
����Jia��(2015)ʹ��֬��ʳ(high fat diet,HFD)ι����C57BL/6JС��ڷ��ܹ���(ursolic acid,UA)����8�ܺ��֣�UAͨ���յ��������ɸ���С��֬�ʺ������Ǵ�л�����۲쵽֬�����ɻ���(��̴�����Ԫ����ϵ���-1(sterol regulatory element-binding protein 1,SREBP1)�µ���Jung��[21]��HFDι��С��ĸ����з��֣�CTRP9�������������յ�AMPK���ữ�����ɣ���SREBP1���������½�������LD�γɡ�Huang��[2]������ʵ����֤����VOdipic-Clͨ���������ɣ���������LD�γɣ�����HFDι��С��Ѫ�����TGˮƽ���ӣ����ٸ���֬�ʻ��ۡ�
����(��)�ٽ�֬�����ģ��ٽ�֬�ηֽ�
����AC2F��ϸ������ʵ��֤����֬�����յ������ɿɴٽ�LD���⣬����С�����֬�ʻ���[19]��Martinez-Lopez��[22]��ʵ��֤���������յ����������ɼ����Ի�����ʽ����֬�����ϸ������֬��ø���������ɿ���ֹ֬�����á�Kaushik��(2015)�����CMAȥ��֬�α���ı������ϣ��ٽ�֬����������װ��ϸ����֬��ø֬�⣬����������ø��֬�ʽ��⡣Martinez-Lopez��(2015)���ֳ�������ϸ������֬��ø����LD�ľ���;���⣬LD������������������:������ѡ������ȡLD����ͨ��������ص�����֬�θ����Ĥ���ʣ�����������ø���ںϺ�������ø��ø�ֽ�LD��֡���������LD�����ںϼ����������¸���֬�ʻ��ۼ�֬������[15]��
�����ٽ�TG��Ա�����ɴ�֬����������ԱTG��Schulze��[15]����С����������øRab7�Ǹ�ϸ��֬�����ɵĹؼ�Э���ߣ�Ӫ����������֬�κ���ø����ұ����Rab7����ٽ�TG��Ա��Zhang��(2015)��ʵ�����ݱ������������ƿ�ͨ��������ǿTG�ֽ⡣Zhang��[23]������֬����M(apolipoprotein M,ApoM)�ó�С���о�����TG��л������ApoMȱʧ�������ɹ����ϰ�����Ҫͨ������TG�ֽ�¸���֬������(TG����)��
�����ٽ�֬��������������ʱ�������ܽ���LD��ʹ֬������������屻������Baena��(2015)��Ϊ�������ɵ���TG����������֬�����γɼ��٣��Ӷ�����β-�����ĵ�����ظ���֬�����ԡ�Tetsuya��[24]���֣���ʳ״̬�����ɴٽ������干��������1(NCoR1)���⣬ʹPPARα����Ӷ��ٽ�β-������ͪ�����ɡ�
�����塢֬�ʴ�л�����ɵ�Ӱ��
�����������о�����������֬�ʴ�л֮����Ӱ�졣������ά��֬�ʴ�л��̬����ؼ����ã�֬�ʴ�л��ƽ�ⷴ�����յ�����������ֹͣ���Ӷ�����֬�ʴ�л��ʹ��ָ���ƽ��״̬���������ijЩ�����缫ǿ����������ʹ���Ϊ�������������١�
����֬������ʱ:Alexaki��(2014)������������֬��л�йأ���������֬��ͷ�ϳɹ��Ȼ�Ծ���յ����ɡ�Nakadera��(2016)���ָ���֬�����Ե���V-ATPø�µ���ͨ���ƻ���ø��������ữ�������ɹ����ϰ���Deng��(2017)���ֹ���֬�ʺ͵��̴���������ϸ�����ɹ��ܣ����ٽ�ERS��HFDһ�������ǿ���ɣ���һ����Ҳ���������ɣ����º��ڵ�֬�ʻ��ۡ�Baena��(2015)����Դ���10%Һ�����8�ܺ��ָ���֬�ʻ��ۿ�������ȱ�ݡ�
����֬�ʼ���ʱ:�ո���������Ӫ������ʱ��Ϊά���������������弤�������Էֽ�֬�ʣ�����LDs������ͨ��ֱ������֬�ʻ���������ʽ���ܡ��������°���������������ڵ�֬������֯�����γ�֬�Σ����ͪ����������Ҫ�����⣬�������������ȱ�ݵĸ��������������Takagi��(2016)�ķ���֤ʵ�˸Ρ������ɶԼ����յ���֬���γɺ�����ͪ����������Ҫ������ά��ȫ��������̬��
�����������:Zhang��(2016)���ָʲ��㶹��(GCM)ͨ��֬�ʴ�л�������¼�����������ɡ�Zhang��(2016)����֧��������BCAA�������m TOR�������֬���յ��ĸ������ɣ��ٽ���ϸ����������ϸ���FFA/TGת���������Ӹ�ϸ����FFA�鵼��֬���Լ����ԡ�֬�ʻ��ۿ��յ����ɣ�BCAA���ƴ������ɣ����ظ������ˡ�
�������������������ڸ���֬�ʴ�л�Ĺ���������Ҫ�ĵ������á���ʳ��������ҩ������ؿ�ͨ����������;�����ڸ���֬�ʴ�л����Щ����ʹ֬�ʴ�л���ң����¼�������;��Щ����ʹ���ҵ�֬�ʴ�л�ָ�ƽ�⡣֬�ʴ�л������֮���Ӱ������ġ��о�����Ӱ�����֬�ʴ�л��;������ΪԤ�������Ƹ���֬�ʴ�л�����ṩ�°е㡣
���������
����[1] Wei CC,Luo Z,Hogstrand C,et al.Zinc reduces hepatic lipid deposition and activates lipophagy via Zn2+/MTF-1/PPARαand Ca2+/Ca MKKβ/AMPK pathways.FASEB J,2018,32�X6355~7028��
����[2] Huang Y,Liu F,Zhang F,et al.Vanadium(IV)-chlorodipicolinate alleviates hepatic lipid accumulation by inducing autophagy via the LKB1/AMPK signaling pathway in vitro and in vivo.J Inorg Biochem,2018,183�X66~76��
����[3] Lee JH,Jia Y,Thach TT,et al.Hexacosanol reduces plasma and hepatic cholesterol by activation of AMP-activated protein kinase and suppression of sterol regulatory elementbinding protein-2 in Hep G2 and C57BL/6J mice.Nutr Res,2017,43�X89~99��
����[4] Chen M,Liu J,Yang L,et al.AMP-activated protein kinase regulates lipid metabolism and the fibrotic phenotype of hepatic stellate cells through inhibition of autophagy.FEBSOpen Bio,2017,7�X811~820��
����[5] Li M,Sharma A,Yin C,et al.Metformin ameliorates hepatic steatosis and improves the induction of autophagy in HFD-induced obese mice.Mol Med Rep,2017,16�X680~686��
����[6] Rujano MA,Serio MC,Panasyuk G,et al.Mutations in the X-linked ATP6AP2 cause a glycosylation disorder with autophagic defects.J Exp Med,2017,214�X3707~3729��
����[7] Zhong J,Gong W,Lu L,et al.Irbesartan ameliorates hyperlipidemia and liver steatosis in type 2 diabetic db/db mice via stimulating PPAR-�AMPK/Akt/m TOR signaling and autophagy.Int Immunopharmacol,2017,42�X176~184��
����[8] Hsiao PJ,Chiou HC,Jiang HJ,et al.Pioglitazone enhances cytosolic lipolysis��β-oxidation and autophagy to ameliorate hepatic steatosis.Sci Rep,2017,7�X9030��
����[9] Sun Y,Xia M,Yan H,et al.Berberine attenuates hepatic steatosis and enhances energy expenditure in mice by inducing autophagy and fibroblast growth factor 21.Br J Pharmacol,2018,175�X374~387��
����[10] Ding S,Jiang J,Zhang G,et al.Resveratrol and caloric restriction prevent hepatic steatosis by regulating SIRT1-autophagy pathway and alleviating endoplasmic reticulum stress in high-fat diet-fed rats.PLo S One,2017,12�Xe0183541��
����[11] Sathyanarayan A,Mashek MT,Mashek DG.ATGL promotes autophagy/lipophagy via SIRT1 to control hepatic lipid droplet catabolism.Cell Rep,2017,19�X1~9��
����[12]֣责����������ף��ȣ�������Ӧ���յ������ɶԸ�ϸ��������Ӱ�죮�й�����������־��2019,35�X332~339��
����[13]�쾲��³����������Ӧ�������ɼ��佻������Ӱ����Ƥϸ���������й����ﻯѧ���������ѧ����2019,35�X240~250��
����[14] Lundquist MR,Goncalves MD,Loughran RM,et al.Phosphatidylinositol-5-phosphate 4-kinases regulate cellular lipid metabolism by facilitating autophagy.Mol Cell,2018,70�X531~544��
����[15] Schulze RJ,Rasineni K,Weller SG,et al.Ethanol exposure inhibits hepatocyte lipophagy by inactivating the small guanosine triphosphatase Rab7.Hepatol Commun,2017,1�X140~152��
����[16] Su H,Yang F,Wang Q,et al.VPS34 acetylation controls its lipid kinase activity and the initiation of canonical and non-canonical autophagy.Mol Cell,2017,67�X907~921��
����[17] Liao X,Song L,Zhang L,et al.LAMP3 regulates hepatic lipid metabolism through activating PI3K/Akt pathway.Mol Cell Endocrinol,2018,470�X160~167��
����[18] Enrich C,Rentero C,Grewal T.Annexin A6 in the liver:From the endocytic compartment to cellular physiology.Biochim Biophys Acta,2017,1864�X933~946��
����[19] Chung KW,Kim KM,Choi YJ,et al.The critical role played by endotoxin-induced liver autophagy in the maintenance of lipid metabolism during sepsis.Autophagy,2017,13�X1113~1129��
����[20] Tsai TH,Chen E,Li L,et al.The constitutive lipid droplet protein PLIN2 regulates autophagy in liver.Autophagy,2017,13�X1130~1144��
����[21] Jung TW,Kim HC,Abd El-Aty AM,et al.Maresin 1 attenuates NAFLD by suppression of endoplasmic reticulum stress via AMPK-SERCA2b pathway.J Biol Chem,2018,293�X3981~3988��
����[22] Martinez-Lopez N,Garcia-Macia M,Sahu S,et al.Autophagy in the CNS and periphery coordinate lipophagy and lipolysis in the brown adipose tissue and liver.Cell Metab,2016,23�X113~127��
����[23] Zhang X,Zhang P,Gao J,et al.Autophagy dysregulation caused by Apo M deficiency plays an important role in liver lipid metabolic disorder.Biochem Biophys Res Commun,2018,495�X2643~2648��
����[24] Saito T,Kuma A,Sugiura Y,et al.Autophagy regulates lipid metabolism through selective turnover of NCoR1.Nature Communications,2019,10�X1567.





