
��������ϸ��Ǩ����������( macrophage migrationinhibitory factor��MIF) �ǵ�һ�������ֵľ��ж�Ч���Խ��ʹ��ܵ�ϸ�����ӣ�ͬʱҲ����Ҫ���ڷ��ڼ��أ����б�λø��������ԭø����ø���ԡ�MIFԤ�Ⱥϳɴ����ڰ����У���������ϸ���絥��/����ϸ����B ϸ����T ϸ�����Լ�������ϸ������Ƥϸ������Ƥϸ�����ڷ���ϸ����Ѫ��ƽ����ϸ���Ⱥϳ�; ������������-����-������Ƥ�����ж��д��棬�Ա����źŴ̼�ʱ�����ͷš�MIF �IJ����ͷ��ܶ������ص��ڣ�����ϸ����л�����ֳ�źš�������������������( �� IFN-γ��TNF-α����Ƥ�ʼ��ص�)��MIF�������ڰ��ڻ��߰��ⶼ������Ӧ�Ĺ��ܣ������������ MIF ��Ѫ��Ũ��Ϊ 2 ��10ng/ml��������ҹ��������
����MIF ���ж��صķ��ӽṹ�����������������кܴ�ͬ���������˽ṹ���� D-��Ͱ���λøͬԴ��Lue ��( 2002) ���� MIF �Ĺ����Է���ΪͬԴ�����壬�䵥��Ϊ 12�� 5kD������ 2 �� α ������ 6 �� β�۵����� MIF �����У�������Ƭ�� 50 �� 68 �� 86 ��102 ��Ϊ��Ҫ�����߶����� β �۵��������е� 57 �� 60λ������Ϊ������ԭ��������λ�㡣����������MIF Ƭ��ͻ����������������ʱ����Ӧ�� MIFø���Խ��ͣ����յ����������ͷŵ������½���
����һ��MIF �鵼���ź�ͨ·
����MIF ��Ϊ��ܵ��������ӣ�����ڰ��ںͰ��ⷢ����Ӧ�Ĺ��ܡ���ˣ�MIF �ȿ���Ϊ�źŷ�������������ϸ�����ֿ�ͨ���Է��ں��Է��ڵ���ʽ����������ں��������ڡ�MIF ͨ���� CXC���塢CD74 �������ϣ�ͨ���� AKT��AMPK��E��K������ͨ·�鵼��������Ӧ�����á�
����( һ) MIF ��������� �� MIF ��ص������
�������� CXC ���������������� CXC��2��CXC��4 ��CXC��7���Լ� CD74 �� CD44����Щ���岢���ǹ����ģ����������帴�������ʽ( ͼ 1) ��� MIF�������źš����帴������� CXC��2/CD74 �������CXC��4 / CD74 �����CD74 ���� MHCII �Ϳ�ԭ���ݵ�������̶���ԭ�����õĿ�Ĥ�ǵ��ף��ް��ڻ��Բ��֣����� CD44 ���ɸ������������źŵ�ת��; CD44 ������ø���ԣ�MIF �� CD74/CD44 �������Ϻ�ʹ����˿�������ữ������ Src �Ұ��ἤøͨ·���鵼�źŴ��ݡ�
������ͬ��ϸ��ϵ�����������Ͳ�ͬ������ϸ��������Ҫ���� CXC��2; T ϸ��������Ҫ���� CXC��4;��ϸ��������Ҫ���� CXC��2 �� CXC��4�����ϵ���Щ������� G ����ż�����塣����� MIF ֻ���� CXC��2 ���ڵ�����£�������ɶ�ϸ������ļ��MIF ����α L�� �ṹ����˿�ģ�� EL�� �ṹ��������Ӧ CXCLs �Ĺ��ܣ��� CXC��2 �����ϡ��о�֤ʵ��CXC��2 �ɽ鵼 MIF �������������ã��Լ���������Ӳ���ȼ���������CXC��4 �� AKT �ź�ͨ·��ء�
����MIF ͨ������鵼���źŴ�����������Ĥ������( plasma membrane receptor) �йأ����������о�����������������( endocytosis of the ligated recep-tor) Ҳ�кܴ��ϵ����������Ҫ�������������У��� MIF ��ص������������������壬��LDL ���̻���������֮����MIF ��ϸ������ CX-C��4 / CD74 �����Ϻ�ͨ���������ý���ϸ�����γ��źŴ����Ͱ����壬�ɲ����źŴ�������һ;���ڵ���ϸ����T ϸ����ϸ��ϵ���з��֡���һ������;���� PI3/AKT �ź�ͨ·�кܴ��ϵ����Ȼ������ AKT ͨ·�������źŴ���;�������ϸ��ϵ�йء�ͬʱ����OS ��ػ���Ҳ�� MIF ������أ�Th1���ϸ�������� IFN-γ��TNF-α��IL-12 �ȿɴٽ� MIF����; Th2 ���ϸ�������� IL-4��IL-10 ����ֹ MIF���̡�
����������Щ�����ź�ͨ·Ϊ����ͨ·��������о���ʾ������ CXC��2/4 ����� CD74 ȱʧ������£�MIF ���źŴ��ݹ��ܽ���Ӧ���ͻ��жϡ����ں��Ƽ�����ϸ��ϵ( ��MS) �У��� CXC��2 �� CD74 �ı���� MIF ����ͨ�� CXC��4 �� CXC��7 �鵼�źŴ��ݣ�����ϸ���������������ӵķ�Ӧ�Լ�ϸ����ֳ��Ѫ��������ͬ�������� CD74 �����������ϸ��ʱ�� MIF Ҳ���Է��ӹ��ܡ���Щ������д��ڽ�һ�����о���
����( ��) �� MIF ��ص��źŷ���ͨ· �� MIF ��ص��źŷ��Ӱ��� Jab1/JNK ͨ·��PI3K ͨ·��Akt��p53��E��K1 /2 ��( ͼ 1 ) �����潫�����Щ��������Ҫ������
��
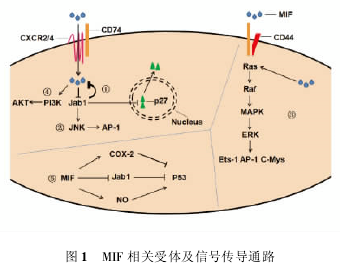
����1�� Jab1: Jab1 ��һ�����������ף��� CSN ���ǵ�λ���� N-ĩ�˺��� MPN �ṹ��MIF �� MPN �ṹ�����ϡ�Jab1 �����ڰ����ںͺ��ڡ�MIF ��Jab1 ���ߵ�Ӱ������ġ�ϸ���Է��ڻ�ͨ��CD74 �����������ϸ����� MIF ����ڵ� Jab1 ֱ�ӽ�ϣ��� Jab1 �з�����ڵ����ã������� Jab1 ��JNK��AP-1 �ȵ����ữ��Jab1 ������� p27 ��ϣ��յ� p27 ���ˣ��ٽ� p27 ����; MIF ����ֹ Jab1�� p27 �Ľ�ϣ����� p27 �ľۼ���ʹϸ������ G1 �ڡ�
����Jab1 �� MIF �Ľ�ϵ��� MIF ����λ�����ڻ����������ư��� MIF �ĺϳɡ�Jab1 Ҳ������ MIF �� Aktͨ·�Ļ���á�Jab1 �Զ����ź�ͨ·���е������ã�����Ե��� β2 �������ź�ͨ·��MAPK �ź�ͨ·�Լ� JNK �ź�ͨ·��
����2�� JNK: JNK �� MAPK �����Ա������˿ / �հ��ᵰ��ø����ø���ܽ�� c-Jun �İ���ĩ�˲����� c-Jun��c-Jun �� c-Fos ��ͬ��� AP-1��AP-1 ��ϸ��������Ҫ��ת¼���ӣ�ͨ�� MAPK �źŴ���ͨ·���AP-1 �ɵ���ϸ������ת�����ϵ��������ӣ����ذ�ϸ��Ǩ�Ƶȡ�MIF �� JNK ���������������дٽ����ã���һЧ��ȡ����ϸ�������͡�
����Jab1 ��ʹ JNK ���ữ���������� AP-1��MIF ��Jab1 �Ľ�Ͽɷ������ Jab1 �����ã�������� JNK�� AP-1 �����ã��ҳʼ��������ԡ�AP-1 ���µ��ᵼ�¶����źŷ����� Bcl-2��p53 �ȵı������µ�����ȱ������£��ļ�ϸ��ͨ����һͨ·�µ�JNK ͨ·���ԣ������µ� bcl-2 �ı���Ӷ���ֹ�ļ�ϸ�����������������ļ���Bcl-2 ���µ���ʹ��BAD ��ϸ�����������½����Ӷ�����ϸ����������һЧ���ڰ�֢�ķ�����չ��Ҳ����Ҫ���á�ͬʱ������ MIF ͨ�� CD74 ���� JNK ��ͨ·: �� T ϸ���� MIF ��ϸ������� CXC��4/CD74 �����ϣ�ͨ�� Src �Ұ��ἤø���ʹ�� JNK ���η��� PI3K �������ʹ�� JNK/c-Jun/AP-1 ͨ·��һ�����١����ݵļ����һ��Ӧʹ�����ε��źŷ�����Ӧ�������� IL-8 �����ϵ��ȡ�
����3�� E��K1 /2: MIF ͨ�� CD74 / CD44 �鵼�ϵ�E��K1 /2 ͨ·�ı����һͨ·��Ҫ�� G ����ż������鵼 ��as-��af-MAPK/E��K kinase-E��K ;��������Ҳ�ɾ����Ұ��ἤø����ͨ·����as ͨ·������ MIF ������ Ets-1��Ap-1 �� c-Myc ���ź�ͨ·��
��������� MIF Ҳ��ͨ�����̽��룬������ڵ� MLCK����Է������á�MIF �� E��K1/2 �ļ����г�������Ϳ��ٶ��ݼ������ֲ�ͬ�Ļ��ơ��ڳ�������ͨ·�У�Jab1 �Ĺ���������� MIF �Ļ��ԣ��谭 E��K�����ữ��ͬʱ��PI3K �����Ƽ�Ҳ�������� MIF ��E��K1 /2 �Ļ������о�������Ѫ���� MIF �����谭��һͨ·�ı�����ڷ����� MIF ����յ���ͨ·�ij������
����E��K �źŷ��ӵ��ϵ��ɴٽ� cox-2��PGs��p53��MSK ��һϵ�з���ˮƽ����ߡ��ڼ���С��ϸ��ϵ�У�MIF ͨ����һͨ·�ٽ� PGE2 �ı����ϵ����Ӷ��յ� cox-2 �ı��������֢��Ӧ������Ҫ���á�ͬʱ��cox-2 ������ p53 ��ϸ���ڵľۼ����Ӷ�����ϸ�������������������߲��방֢����������Ҫ���á�E��K1/2 ͨ·�����дٽ�Ѫ�����������ã���ͨ���� IL-8 �� MSK1 �� ��SK1 ���������ӵ��ͷţ�������ϸ�����ԡ�
�����ڶ��ݿ��ټ���ͨ·�У������� Elk-1 ת¼���ӵı����������ϵ���Ѫ���� MIF ������ E��K �ļ�����Ч��ϵ�����Σ������¶ȡ�pH �ȵ�Ӱ�졣��������ݰ��� MIF �������� E��K ͨ·����ټ�����ڳ�������ͨ·���Dz��������ġ��� Jab1�� E��K1/2 ���ϵ��DZ���ġ����ʵ��֤ʵ�ڶ��ݿ���ͨ·�� MIF ͨ���Ұ��ἤø�����źţ���ʹMEK1 /2 ���ữ����Ӧ�������μ�ø ��af-1 ��������������ȫ�����һͨ·��˵������ MEK1/2 ���ε�������ø��������һͨ·�ķ�����
����4�� PI3K / Akt: MIF ͨ���� CD74 ����Ľ��ʹ�� Src �Ұ��ἤø���ữ���Ӷ����� PI3K; PIP3 �������ϵ�����ļ˿/�հ��ἤø Akt ��ϸ��Ĥ�����ữ�������ǰ��ڻ��ǰ���� MIF ���ɵ��� Akt �ı��Akt ����Ĥ����������������崫���źţ����������Ҫ����ϸ��ϵ������Akt ͨ·�� MIF �Ŀ�ϸ�������Լ�Ѫ��������������ء�PI3K �ɴٽ� IL-8��ICAM-1��VCAM-1 ��ϸ�����ӵ��ͷţ��Ӷ��ٽ�Ѫ������; ͬʱ��ICAM-1 ������ϸ����ճ���붯������Ӳ���ķ���Ҳ�кܴ��ϵ��Akt ��ʹBAD �������ữ���Ӷ�ʹ�䲻����ֹ Bcl-2 �� Bcl-xL �������Ĺ��ܡ�ͬʱ Akt ͨ·�ļ���������� caspase-3 �Ļ������ϸ�����������ڰ�֢�ķ����Լ��������кܴ����ʾ���á�
����5�� p53 ����������: MIF �� p53 �Ĺ�ϵ�dz����ӣ�Ҫ����ϸ�������Զ�����Jab-1 ���� p53 ��ϣ��յ� p53 ����ϸ�����н��⣬MIF �� Jab-1 �Ľ�Ͽ����� p53 ����; ͬʱ��MIF Ҳ���յ� NO �IJ������Ӷ��ٽ� p53 �IJ������ٽ�ϸ�����������ǣ�MIF Ҳ���谭 p53 �������ٽ��併���Ч����MIF �ĵ� 81 λ���װ������ p53 ��ϣ��鵼 p53 ����; ͬʱ��MIFҲ��ͨ���� cox-2 �ĵ������� p53 �ۼ����� p53 Ҳ������ MIF �Ĺ��ܡ�
����MIF �����ϵ� TL��-4 �ı��MIF ��ͨ��������һ��ת¼���� ETS �����Ա PU�� 1 ������ TL��-4�Ļ�����ɱ����� LPS ��ϵ����帴���TL��-4 �����źŴ������ܣ������� NF-κB �Ļ��ԣ����������ϸ������������ͷ���֢���ӣ�������ڹ�������������Ҫ���á�MIF ���ϵ����ʽ�������ø( MMPs) �Ĺ��ܣ���һ���ػ�����ͨ�� MEK1/2-JNK( AP-1) -MMPs ͨ·���У������Ǿ��� MMPs �����ͨ·���ù����방ϸ����������Ӳ���߿鲻�ȶ����Լ���������������Ҫ��ϵ��MIF �� NF-κB�����γ����Ի�·���������������� TNF-α��IL-1β��oxLDL�ȿɾ��ɴ�ͨ·�ٽ� MIF ���ͷš�MIF ͨ�� CD74/CD44 ����鵼������Akt ͨ·���� NF-κB ���ͷš�NF-κB �ɵ��ڶ����������ӵı�����������յ� IL-1β��IL-8 ���ɣ���֤ Bϸ �� �� �� �� ��ͬ ʱ MIF Ҳ �� �� �� NF -κB �� p53֮��Ĺ�ϵ����ϸ��˥������֢��Ӧ������Ҫ���á�
�����������о����Կ�����MIF ���ͨ·��Ĺ�ϵ�����ǹ����ģ������Ե����������ʽ���ڡ�MIF�Ե�һ���źŷ��ӵĵ���ҲҪ���ݲ�ͬ��ϸ��ϵ������Ŀǰ��MIF �Ļ����о���δ��ȫ�������Ҫ��һ����̽��֤����
��������MIF ����
����MIF ��Ϊ�����������ӣ������༲�����л�Լ���( �綯������Ӳ��) ���������߲�����֢����Ⱦ�Լ���( ���Ѫ֢) �Լ��˿������ж��зdz���Ҫ�����ã���˿���Ϊ��Щ���������������ݴ˰е��о��µ����Ʒ�����ͬʱ��MIF Ҳ����Ҫ���ڷ��ڼ��أ����е���Ѫ�ǡ�������Ƥ�ʼ���( GCs) �Ĺ��ܡ����ڽ����� MIF �Ļ��������Լ����ڸ���Ҫ�����еĹ�������ϸ���ܡ�
����( һ) MIF �Ļ�������
����1�� �뼤�ص������: GCs ����������������Ƶ����ã��ô��ǿ�����ǿ��������ٶԽ��š����Դ̼�����������; �����ǽ������������������Ƶ����ʺϳɵ�һϵ�����⣬Ũ�ȹ��߿ɵ��¿���ʽ�ۺ�֢��MIF �� GCs �����������ġ���Ȼ GC ������ϸ���������������Ӿ����������ã��������MIF �ͷŵĴٽ�����Ҫ����������; �෴ MIF ��GCs ���������ã����� GCs Ũ��ʹ�䲻�¹��ߡ�
����MIF ���ڽ����Լ����������°��� GCs ���ͷţ���ÿ�� MIF �� GCs �����ڵ�Ũ�������кܺõ����϶ȡ�����Ũ�ȵ� GCs �ɴ̼� MIF ���ͷţ����Ǹ�Ũ�ȵ� GCs ������ MIF �ͷš�MIF ͨ���� CD74/CD44 �����ϣ��Ӷ��鵼 MAPK ����ͨ·��ͨ������֬ø A2��������ϩ��ĵ��ڣ��Ӷ��� GCs ���á���Ȼ MIF �� GCs �����߹�����������ϵ�����Ƕ��ߵľ�ȷ����Ҳά�������ߵ�ƽ�⡣������MIF �� GCs ����Ҫ��ϵ�����ڿ���ʽ�ۺ�֢�����ȵ��ع�������ĵ�Ѫ�ǻ����У�MIF �ı�������û����ߣ��������д����о���
����MIF �ɵ����ȵ��ص��ͷ��Լ��Ǵ�л��MIF �����ȵ� β ϸ���ϳɣ������ȵ����ڹ�ͬ���ͷſ������ͷ���Ѫ���������Է��ں��Է��ڼ��ع��ܡ����ֲ�����������ˮƽ����ʱ���ɴٽ� MIF ���ͷţ��Ӷ��ٽ��ȵ����ͷţ�����Ѫ�Ǻ� MIF �ĺ�����
����MIF �ɴٽ����������ļ��������ǵ���ȡ�����á��ڷ���ˮƽ�ϣ�MIF �ϵ�������ת���� 4 ��������Ǽ�ø 2 ��Ũ��; ͬʱ���ͨ���ϵ� TNF-α �ٽ��ǽͽ⣬��һЧ����Ӧ������¸����ԡ���ȫ�����Է�Ӧ���µ�Ѫ�Ǹı������У�MIF ���ȵ������� 1 �� Akt ͨ·�йء�
����2�� ����������: MIF �Ƕ�ܴ��������ӣ�ͬʱ��ɵ��ڹ������������������ߡ�MIF �ɴٽ�����ϸ���Ļ���Ӷ��γ����Է�����·��MIF ͨ������;�������֢ϸ������ļ: ͨ���ϵ� VCMA-1 ��ICAM-1 �����ٽ���ϸ����Ѫ����Ƥճ��; ͨ����CXC��2 /4 �����ϣ��Ӷ��ٽ����Բ�λ��ϸ������ļ; ͨ���ٽ�����ϸ����������-1( MCP-1) �ı��ͬʱ��MIF ͨ���ٽ� cox-2������ p53 ͨ·�Ӷ���ֹ����ϸ����µĵ�����ά�����Է�Ӧ��MIF �Ĵ����Թ��ܿ��� TL��4 ���ڣ���ɴٽ� IL-6��IL-1β��TNF-α ��һϵ���������ӵ��ͷţ�������MAPK ͨ·����������ػ����ת¼���ӣ�ͬʱ����������( ����Ƥ�ʼ���) �����á�
����( ��) MIF ����ؼ����еĹ���
����1�� MIF �ڶ�������Ӳ���е�����: �����о��Ľ�չ�����Ƿ��ֶ�������Ӳ����������֢��ʮ����Ҫ�Ĺ�ϵ�����ܶ�֬����( LDL) ��Ѫ����Ĥ�ij��������ڴٽ���Ƥϸ����ƽ����ϸ������������ MIF��Ϊ�����������ӣ����� LDL �Ĺ�ϵʮ�����У��ɴٽ���������Ӳ���ķ����뷢չ����������Ѫ���У������� MIF ����; Ȼ�������Բ�λѪ���У�MIF �ı����������ϵ���MIF ��Ҫ��Ѫ����Ƥϸ��������ϸ����T ϸ����ƽ����ϸ�����ڡ���Щϸ������֢��Ӧ֮ǰ���ɴ��� MIF���ܵ��̼����� MIFת¼��δ��ʼ�����ͷ� MIF����Щϸ���ڷ���MIF ��ͬʱ�������ٷ���һ�� MIF �����壬�� CD74��CXC��2��CXC��4 �ȣ�������Щϸ���������� MIF �Ĵ��桢���ڴ�л�أ������� MIF ������ϸ��������MIF ���źŴ̼���
����MIF ������Ӳ���ĸ���ʱ�ھ��б���ٽ��䷢չ���о��������� MIF ȱ������ģ���У��߿�Ĵ�С����֢�̶Ⱦ��м��͡�ͬ����Ӧ�� MIF ���кͿ���ɼ��ٰ߿����ˣ�����Ѫ�ܵ���խ�̶ȡ���� MIF �ڼ��Թ����ۺ������������ಡ������Ҫ���á�
�����ڰ߿��У�TNF-α��IFN-γ��CD40L��Ѫ�ܽ����� II( AngII) �Լ��������ܶ�֬����( oxLDL) �ȿɴٽ�MIF �ķ��ڣ������� oxLDL �� MIF ���ͷż���������Ӳ�����γɶ��о����Ե����á��ڰ߿��е���/����ϸ����ļ���� MIF ��ء�MIF ��ͨ����������;���յ���ϸ������߿�: ( 1) �յ�Ѫ����Ƥϸ���� ICAM-1 �� CCL-2 �ĺϳ�; ( 2) MIF Ҳ�����������ӵĹ��ܣ����뵥��ϸ���ϵ� CXC��2 ���壬�� T ϸ������ CXC��4 ������; ( 3) MIF ���ϵ�Ѫ����Ƥϸ��Ĥ�������ء��ڰ߿��γɵ�֬���ڣ�MIF��ɴٽ�������ϸ����������Ƥϸ������������Ӳ�����ڰ߿���γ�����Ҫ���á�����߿��ڵĵ���ϸ������ת��Ϊ����ϸ��������ת��Ϊ��ĭ״ϸ����
�����߿��ڵ� MIF �ɴٽ����������� TNF-α��IL-1β��iNOS��NO �ȵ��ͷţ����Ӱ߿�ֲ��������ã�����Ϊ��ϸ���Ľ�һ����ļ�춨������MIF ������p53 �Ĺ��ܣ��Ӷ��ٽ���֢��Ӧ������ϸ��������p53��ȱʧ�ɵ��¶�������Ӳ���߿���Է��γɡ�
����MIF �ɴٽ��߿���ƽ����ϸ�����ʽ�������ø( MMPs) ���ͷţ����� MMP1��MMP9 �� MMP12 ���Ե��°߿�Ļ���������ѵ�Σ�ա�ͬʱ��MIF ����Ѫ��ƽ����ϸ���ͷŵ�ѪС�������������ӣ��Ӷ���ֹ��߿���ȹ����á�
����Ŀǰ���Ѿ�ӵ������� MIF �������������������ٴ�Ч�������о�֮�С���������о��� MIF ������������һЩ������: ( 1) �����MIF ����λ��; ( 2) �乹����; ( 3) �������� MIF ����λ��л�; ( 4) ͨ���� MIF ����-�����廥���칹���ƻ�; ( 5) �ȶ� MIF ���壬��ֹ�����л��Ե�������ת����
����2�� MIF ���������߲��еĹ���: �� MIF ��ص��������߲�������ʪ�Թؽ��ס�ϵͳ�Ժ���Ǵ�( SLE) �ȡ�����о��������ڷ�ʪ�Թؽ����У���������ѪҺ��ؽڻ�Һ�� MIF �ĺ�������������������֢��λ�Ĺؽڻ�Ĥ�ʡ���Ĥ�ҵ���֯�� MIF �����������������������뼲�����������س̶ȳ�����ء�Ӧ�ÿ� MIF ���������ڷ��Ĺؽڲ�λ����Ӧ������������ TNF-α �ȵĺ����½�������֢������λ��MIF �ܹ���� VEGF( ��Ƥϸ����������)�ĺ�������˾��дٽ�Ѫ�����������á��о��������ڷ�ʪ���У�MIF ���ӹ������¶�ǽ����������ء�MIF �����������������е�����ʮ�־�
������ IgA ��������Ѫ���������Ļ����У�MIF ���������ϵ���MIF �����Դٽ���Ѫ����Ƥ�Ļ���Ӷ��ٽ��������༲���ķ�����
�����о�����������θ���У�MIF ���ͷ�������Ӧ����أ���������IJ���̶��أ�ͬʱ������������Ѫ�ܼ�����ء�MIF ͨ��� T ϸ���յ����༲���������� MIF ��ļ�� T ϸ�������࣬�յ���֢��Ӧ; MIF ���յ� T ϸ������ֳ�ͻ��ͬʱ MIF ��ͨ���̼����������յ�����ϸ���; MIF ����Ƥ�ʼ��ص��������ü�����������֢����������ˡ�
����MIF ����ϵͳ�Ժ���Ǵ�( SLE) �и߱������һԭ���� GCs ������ء����ڸ�Ũ�� MIF�� SLE �����У���Ԥ���벢��֢�������������Ѫ�� MIF ���ļ����Ժܺõ����� SLE �����س̶ȡ�
����MIF �鵼�� SLE ������ CD74 / CD44 ����鵼��B��T ϸ����������ɵġ�GCs ����Ϊ SLE ���Ƶľ��䷽�������ǽ����м����о����� GCs �� SLE ��Ԥ���в������ã�����һ����� MIF �кܴ��ϵ��
����GCs ͨ���ϵ� IF-κBα ���״Ӷ����� NF-κB �Ļ��ԣ�������������; �� GCs �ֿɼ��� MIF ���ͷţ�MIF�ɼ��� NF-κB ��·���Ӷ��յ� GCs �ֿ����ٽ���֢��Ӧ������ʹ SLE �IJ�������ڸ��������Ƥ�ʼ������ƵĻ�����˵���鿴 MIF ����̴���ˮƽ��ϵ������ϻ����Ƿ���DZ�����Է�Ӧ�ķ��գ��Ի������� MIF ˮƽ�����ж��������Ƿ����ۡ�
����3�� MIF �ڰ�֢�е�����: ��֢�ķ�������������йأ�����������������������أ�����������֢��֢�����Ĵ̼������Ѿ�������֤ʵ����֢�������������ص�: �����յ���ֳ����; �Կ���ֳ���źŷ��Ӳ�����; ����������; DZ�ڵ�������ֳ����; �ٽ�Ѫ������������; �Լ���Χ��֯����ת�Ƶ��������о�֤ʵ MIF �ڶ��ְ�֢�о��б���� MIF��ϸ���������ص㶼��һ���Ĵٽ����ã��Ӷ��ٽ��������ķ�����MIF �����ٰ��Ķ����к���Ҫ�����á�MIF ͨ���� p53 �Ĺ��ܴӶ���ֹ��ϸ���ĵ���; ͨ���ٽ� E��K1/E��K2 �ļ����ź�ͨ·���Ӷ��ٽ� NOS2 �� NF-κB ͨ·��������ֹ p27 �Ľ��⣬�Ӷ�ʹ��ϸ���ֻ��̶ȸ��ͣ����Գ̶ȸ����ԡ�
����ZEB1 /2 �ɴٽ���Ƥ���ʻ��� ( EMT) ���Ӷ�����ϸ���ֻ��̶ȵͣ��ٽ���������; �� mi��-200b ����ZEB1 /2 ��ϣ��γɸ�������·������ ZEB1 /2 ���ԣ��γɼ�����Ƥ����( MET) ��MIF �ɴٽ� ZEB1/2 �Ļ��ԣ����� mi��-200b �Ļ��ԣ����� E-��ճ�صĺ�������߲��ε��Ļ��ԣ��Ӷ��������ٰ�ϸ����Ƥ���ʻ�����ͬʱ��MIF �������ٰ�ϸ���Ľ�������������ء�MIF ����Ϊ���ٰ���������Ϻ�����Ԥ���ָ�ꡣ
��������о��������ڹ��������ܰ�ת�ƹ����У���ͬ���͵� MIF ��ϸ���ܰ�ת�ƵĴٽ����ò�ͬ���� 173 λ������ͻ��Ϊ����ऺ����֢�Ĵٽ����ü��ܰ�ת�ƶ������ǿ������һЧ����Ұ����MIF ��û�����֡�MIF ��֢�ķ�����չ�Ĵٽ����û��кܶ�棬�����ﲻһһ�о١�
����4�� MIF �ڴ�л�Լ����е�����: MIF ��ͨ���伤�������ã����ȵ��ؼ���Ƥ�ʼ��صȽ��е��أ��ɵ��µļ����������֡�������������Ӳ����Ŀǰ�����ֻ��������࣬�������ữ���ơ��о�������MIF �ɴٽ����������֢�ķ������� BMI �Ļ����� MIF �����ߣ�������Ѫ�� FFA ��л���� C��P ��ء�MIF �����ķ�������չ����Ҫ���á��������� 2 ������������Ҫ���أ��о�������MIF ���������ķ���������Ҫ���á�MIF �鵼��TNF-α ��������ʹ���ȵ��صֿ�����һ������ Aktͨ·�鵼���Ӷ��ٽ������ķ�����MIF ���ȵ�Ҳ�����������ã����ʹ����������ϸ�����࣬���յ����ȵ�ϸ�����𣬴ٽ� 2 �����ķ�������ͨ·�� CXC��4 ���弰 G ����ż������ 2 ��ø�鵼������Ҳ������ʵ�������MIF ��ȱʧ����ֹ���������� INF-γ��TNF-α��IL-1β �ȵ����ã��Ӷ������ȵ�ϸ�����ڵ���; ��������ͨ���µ����������ͨ·��E��K1/2 ͨ·�� NF-κB ͨ·��ɵġ�
����5�� MIF �����������е������½�չ: MIF ���յ��ķ��˲���T ������ͨ��ȱʧ���ķ��˲���������Ҫ���ơ����� MIF �Ը�����ͨ���Ĺ��ܣ�MIF��ͨ���Ұ��ἤøͨ·���� T ��ͨ���� α1G ��α1H �ǵ�λ���Ӷ��յ��ķ��˲��ķ�����MIF �ɸ������������ơ�MIF ������ϸ���з��ڣ��俹�����Ĺ����ڽ��ڱ����֡�Ŀǰ�������֢�ķ�������������ɫ��ѧ˵���Ȱ���ѧ˵�ȣ���Ϊ���ϵķ�������������������������ɫ���ϳɼ��ٵ���ɫ�����ǻ�ø 2( Tph2) ������Ӫ������( Bdnf) ��������Կ������������ƺ����ߵ���������ָ�ꡣ
������������� MIF �ı�����������������ָ������������ͬʱ������ Dcx �� Pax6 ��Ũ��Ҳ����������֤������������Ԫ�ķ�����MIF ���������������ɫ��Ũ�ȵĹ��ܣ������ǿ��Ʒ��͵���Ʒ��ȿ��������Ƶ�Ч����MIF ����һ�����Ǿ���CD74 / G ����ż�����塢��hoA-E��K1 /2 ͨ·�鵼�ģ�Ӧ�� MIF �����������ͨ·���ͼ����谭���������Ƶ������ԡ�
��������չ��
�������Ŷ� MIF �о����ս����룬���书�ܵ��˽�Ҳ���˽�һ���ķ�չ������ٽ���������ؼ������˽⣬ҲΪ�°е�����Ƶ춨�˻�����Ŀǰ���MIF �Ĺ����Ѿ����˲����ٴ���Ӧ�ã�����Ϊ��ؼ�������ϱ�־�����Ϊ��ؼ��������ưе㡣���Ƕ��� MIF ���о���û����ȫ�������Ӧ�÷�Χ���н�һ��������ռ䣬�д������ǽ�һ�����о���
������ �� �� ��
����1 Simons D��Grieb G��Hristov M��et al�� Hypoxia �\ inducedendothelial secretion of macrophage migration inhibitory fac-tor and role in endothelial progenitor cell recruitment�� J CellMol Med��2011��15�X668 �� 678��
����2 ��osado Jde D����odriguez-Sosa M�� Macrophage migration In-hibitory factor ( MIF) : a key player in protozoan infections��Int J Biol Sci��2011��7�X1239 �� 1258��
����3 Grieb G��Merk M��Bernhagen J��et al�� Macrophage migra-tion inhibitory factor ( MIF) : a promising biomarker�� DrugNews Perspect��2010��23�X257 �� 264
����4 Lue H��Thiele M��Franz J��et al�� Macrophage migration in-hibitory factor ( MIF) promotes cell survival by activation ofthe Akt pathway and role for CSN5 / JAB1 in the control ofautocrine MIF activity�� Oncogene��2007��26�X5046 �� 5059��
5 Bernhagen J��Krohn �ң�Lue H��et al�� MIF is a noncognateligand of CXC chemokine receptors in inflammatory andatherogenic cell recruitment�� Nat Med��2007��13 �X 587 ��596��
����6 Schwartz V��Krüttgen A��Weis J��et al�� ��ole for CD74 andCXC��4 in clathrin-dependent endocytosis of the cytokineMIF�� Eur J Cell Biol��2012��91�X435 �� 449��
����7 Lue H��Dewor M��Leng L��et al�� Activation of the JNK sig-nalling pathway by macrophage migration inhibitory factor( MIF) and dependence on CXC��4 and CD74�� Cell Signal��2011��23�X135 �� 144��
����8 Gesser B����asmussen M����aaby L��et al�� Dimethylfumarateinhibits MIF-induced proliferation of keratinocytes by inhibi-ting MSK1 and ��SK1 activation and by inducing nuclear pc-Jun ( S63 ) and p-p53 ( S15 ) expression�� Inflamm ��es��2011��60�X643 �� 653��
����9 Sivaram G��Tiwari SK��Bardia A��et al�� Macrophage migra-tion inhibitory factor��Toll-like receptor 4��and CD14 poly-morphisms with altered expression levels in patients with ul-cerative colitis�� Hum Immunol��2012��73�X201 �� 205��10 Salminen A��Kaarniranta K�� Control of p53 and NF-κB sig-naling by WIP1 and MIF: role in cellular senescence andorganismal aging�� Cell Signal��2011��23�X747 �� 752��
����11 Miller EJ��Li J��Leng L��et al�� Macrophage migration in-hibitory factor stimulates AMP-activated protein kinase inthe ischaemic heart�� Nature��2008��451�X578 �� 582��
����12 Emonts M��Sweep FC��Grebenchtchikov N��et al�� Associa-tion between high levels of blood macrophage migration in-hibitory factor��inappropriate adrenal response��and earlydeath in patients with severe sepsis�� Clin Infect Dis��2007��44�X1321 �� 1328��
����13 Amin MA��Volpert OV��Woods JM��et al�� Migration inhib-itory factor mediates angiogenesis via mitogen-activated pro-tein kinase and phosphatidylinositol kinase�� Circ ��es��2003��93�X321 �� 329��
����14 Amin MA��Haas CS��Zhu K��et al�� Migration inhibitoryfactor up-regulates vascular cell adhesion molecule-1 andintercellular adhesion molecule-1 via Src��PI3 kinase��andNFκB�� Blood��2006��107�X2252 �� 2261��
����15 Veillat V��Lavoie CH��Metz CN��et al�� Involvement of nu-clear factor-κB in macrophage migration inhibitory factorgene transcription up-regulation induced by interleukin-1βin ectopic endometrial cells�� Fertil Steril��2009��91 �X2148 �� 2156��
����16 Cao W��Morin M��Sengers V��et al�� Tumour necrosis fac-tor-α up-regulates macrophage migration inhibitory factorexpression in endometrial stromal cells via the nuclear tran-scription factor NF-κB�� Hum ��eprod��2006��21 �X 421 ��428��
����17 Benigni F��Atsumi T��Calandra T��et al�� The proinflamma-tory mediator macrophage migration inhibitory factor in-duces glucose catabolism in muscle�� J Clin Invest��2000��106�X1291 �� 1300��
����18 Atsumi T��Cho Y�ң�Leng L��et al�� The proinflammatorycytokine macrophage migration inhibitory factor regulatesglucose metabolism during systemic inflammation�� J Immu-nol��2007��179�X5399 �� 5406��
����19 Weber C��Noels H�� Atherosclerosis: current pathogenesisand therapeutic options�� Nat Med��2011��17�X1410 �� 1422��20 Tillmann S��Bernhagen J��Noels H�� Arrest functions of theMIF ligand / receptor axes in atherogenesis�� Front Immunol��2013��4�X115��
����21 Verschuren L��Kooistra T��Bernhagen J��et al�� MIF defi-ciency reduces chronic inflammation in white adipose tissueand impairs the development of insulin resistance��glucoseintolerance��and associated atherosclerotic disease�� Circ��es��2009��105�X99 �� 107��
22 Herder C��Illig T��Baumert J��et al�� Macrophage migrationinhibitory factor ( MIF) and risk for coronary heart disease:results from the MONICA / KO��A Augsburg case-cohort��study��1984-2002�� Atherosclerosis��2008��200�X380 �� 388��
23 Müller II��Müller KA��Sch��nleber H��et al�� Macrophagemigration inhibitory factor is enhanced in acute coronarysyndromes and is associated with the inflammatory re-sponse�� PLoS One��2012��7�Xe38376��
����24 Lin SG��Yu XY��Chen YX��et al�� De novo expression ofmacrophage migration inhibitory factor in atherogenesis inrabbits�� Circ ��es��2000��87�X1202 �� 1208��
����25 Gregory JL��Morand EF��McKeown SJ��et al�� Macrophagemigration inhibitory factor induces macrophage recruitmentvia CC chemokine ligand 2�� J Immunol��2006��177 �X8072 �� 8079��
����26 Ouertatani-Sakouhi H��El-Turk F��Fauvet B��et al�� Identi-fication and characterization of novel classes of macrophagemigration inhibitory factor ( MIF) inhibitors with distinctmechanisms of action�� J Biol Chem��2010��285 �X26581 ��26598��
����27 Xu L��Li Y��Sun H��et al�� Current developments of macro-phage migration inhibitory factor ( MIF) inhibitors�� DrugDiscov Today��2013��18�X592 �� 600��
����28 Col-Araz N��Pehlivan S��Baspinar O��et al�� Association ofmacrophage migration inhibitory factor and mannose-bindinglectin-2 gene polymorphisms in acute rheumatic fever�� Car-diol Young��2013��23�X486 �� 490��
����29 Wang FF��Zhu LA��Zou YQ��et al�� New insights into therole and mechanism of macrophage migration inhibitory fac-tor in steroid-resistant patients with systemic lupus erythe-matosus�� Arthritis ��es Ther��2012��14�X��103��
����30 Funamizu N��Hu C��Lacy C��et al�� Macrophage MigrationInhibitory Factor ( MIF) induces epithelial to mesenchymaltransition��enhances tumor aggressiveness and predicts clin-ical outcome in resected pancreatic ductal adenocarcinoma��Int J Cancer��2013��132�X785 �� 794��





