
引言
【研究意义】菊花(Chrysanthemum×morifoliumRamat.)是中国传统名花,深受人们的喜爱。在世界范围内,菊花是市场上主要的鲜切花之一,也是重要的盆花、地被花卉。菊花起源于中国,是菊属部分野生种天然杂交再经人工选育形成的,其主要亲本为毛华菊(C.vestitum Hemsl.)、野菊(C.indicum L.)与紫花野菊(C.zawadskii Herb.)等,经过长期天然杂交、人工选择和培育,产生了大量的种下变异,具有丰富的遗传多样性。菊花的表型性状大部分是数量性状,变异极其丰富,因其极易受环境因素的影响,多年来对菊花复杂性状的研究始终极为困难。对菊花重要表型性状的基因型进行解析,寻找与目标性状相关的分子标记,将为复杂数量性状的遗传学研究、品种鉴定及品种保护和分子标记辅助育种奠定重要基础。【前人研究进展】分子标记技术已成功应用于菊花的起源研究、品种鉴定和遗传多样性分析等领域。随着分子标记技术的发展,利用全基因组的分子标记和合适的分离群体寻找连锁标记、开展遗传连锁作图是目前植物数量性状研究的主要方法之一。菊花长期进行无性繁殖,基因组高度杂合,且存在近交衰退现象,所以,菊花的遗传图谱构建一直是个难题,在此方面的研究工作刚刚起步。赵婧媛等利用地被菊和盆栽小菊的杂交 F1代群体,通过集团分离分析(bulked segregant analysis,BSA)法寻找到与菊花匍匐性显着相关的 RAPD 标记。Zhang 等根据“双-假测交”作图策略,利用两个高度杂合的菊花品种作为亲本,将其杂交 F1代作为作图群体,使用RAPD、ISSR、AFLP 和 SRAP 等分子标记成功构建了双亲的遗传连锁图谱,并筛选出与菊花初花期和开花持续期相关的 SRAP 标记,还对花径、舌状花数等花器性状进行了数量性状位点(quantitative trait loci,QTL)定位。尽管部分连锁图已经在菊花中建立起来,但是标记密度较低、连锁群不完整,制约了其应用潜力。【本研究切入点】关联分析,又称关联作图或连锁不平衡作图,曾广泛用于人类遗传学研究,是一种研究复杂数量性状的遗传学方法。它是以自然群体为研究对象,以长期重组后保留下来的等位基因(位点)间连锁不平衡(linkage disequilibrium,LD)为基础,将目标表型性状的多样性与基因(位点)的多态性结合起来分析,可直接鉴定出与表型变异密切相关且具有特定功能的基因位点或标记位点。与传统的连锁作图相比,关联分析有以下优点:以自然群体为材料,无需构建作图群体;可以同时检测同一座位的多个等位基因,而连锁作图只能涉及来自亲本的2 个等位基因;广泛的遗传材料可同时考察多个性状的大多数 QTL 的关联位点及其等位变异,不受连锁作图中两亲本范围及群体分离情况的限制。鉴于利用连锁作图开展菊花园艺性状 QTL 定位的难度大、周期长,因此可以考虑利用关联分析的特点,充分发挥中国菊花种质资源丰富多样的优势,以自然群体为基础开展关联分析,鉴定与重要表型性状关联的分子标记,从而为菊花分子辅助育种服务。【拟解决的关键问题】本研究在分析群体结构的基础上,用关联分析对 58 个大菊品种的 18 个数量性状与 SRAP 分子标记进行关联,希望鉴定出与上述性状相关联的标记位点,为菊花育种工作的深入开展和品种鉴定提供一些有价值的参考资料。
1 材料与方法
1.1 供试材料
本研究所有材料均来自北京林业大学菊花资源圃。在 2008—2010 连续 3 年对菊花资源圃内 800余个大菊品种表型性状进行观测分析的基础上,以尽可能覆盖大菊品种出现的所有性状变异为原则,在资源圃中选择具有代表性的 58 个大菊品种(涵盖 5 个瓣型,30 个花型;包括 50 个中国品种和 8个日本品种),为无直接亲缘关系的自然群体(表1)。
所有品种于 5 月在日光温室中扦插繁殖,6—7 月定植于直径 30 cm 瓦盆中,按照独本菊的栽培技法常规田间管理。
1.2 方法
1.2.1 表型性状的测定与分析 本研究中测定 18 个数量性状,包括:花部性状 11 个(花径大小、花瓣长度、花瓣宽度、花梗长度、花高、花梗粗度、舌状小花数、筒状小花数、筒状花长度、筒状花部直径、花托大小);叶部性状 4 个(叶柄长度、叶片长度、叶片宽度、叶厚);茎部性状 2 个(节间长度、茎粗);整体性状 1 个(植株高度)。性状的测定方法参照雒新艳等的描述。每个品种测量 10 个单株,取平均值。计算各性状在品种内和品种间的变异系数,计算公式为:CV=S/X,S={[EX2-(EX)2/N]/N-1}1/2其中,CV 为变异系数,S 为标准差,X 为样本均值。
1.2.2 DNA 提取 田间取植株嫩叶,用改良 CTAB 法提取基因组 DNA。用 1%琼脂糖凝胶电泳和紫外分光光度计测定 DNA 质量和浓度,并将浓度稀释至 50ng·μL-1,样品在-20℃下保存备用。
1.2.3 SRAP 分析 选用正向引物 13 条,反向引物 10条,共 130 对 SRAP 引物用于引物筛选。SRAP 分析参照张飞等的方法,经过体系优化及引物筛选后,对供试品种进行扩增。将具有相同迁移率的扩增片段,按照二进制方法进行记录,即有带的记为 1,无带的记为 0,仅记录清晰、稳定的扩增条带,最终形成 0/1矩阵输入计算机。遗传多样性分析采用多态性信息量(Polymorphism Information Content,PIC)指标。按照 Anderson 等的计算方法,标记 i 的 PIC 值计算公式为:【1】
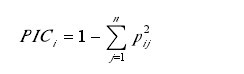
其中,pij表示标记 i 第 j 种带型出现的频率,标记 i的带型数从 1 到 n。PIC 值的范围为 0—1,0 表示无多态性,1 表示具有非常高的多态性。
1.2.4 数 据 统 计 与 分 析 群 体 结 构 分 析 用STRUCTURE软件对菊花品种群体结构进行基于数学模型的类群划分。先设定群体数目(K)为 2—9,将 MCMC(Markov chain Monte Carlo)开始时的不作数迭代(Length of burn-in period)设为 10 000 次,再将不作迭代后的 MCMC 设为 100 000 次,然后计算出每个 K 值对应的 lnP(D)值,根据似然值最大的原则,选择合适的K值,并计算得到每个品种相应的Q值(第i 品种其基因组变异源于第 k 群体的概率)。关联分析应用 TASSEL 软件 GLM(General Linear Model,一般线性模型)程序,将各个菊花品种的 Q 值作为协变量,将18个数量性状的表型性状数据分别对SRAP标记变异逐一进行回归分析,确认表型性状关联位点并计算位点对表型变异的解释率。GLM 回归方程是:【2】
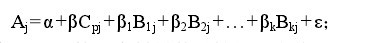
其中,Aj是第 j 个材料数量性状测定值,β 是群体各位点各等位变异的平均效应,Cpj是第 j 材料第 P 等位变异出现的指示变量,B1j—Bkj是第 j 个材料基因组变异源于第 l—k 群体的概率 Q 值,β1—βk是亚群体各位点各等位变异的平均效应,ε 是残差。
2 结果
2.1 数量性状的变异分析
本研究中应用变异系数来表示观测的数量性状在品种内和品种间的离散性程度。品种内变异系数反映了性状在同一品种内的稳定程度,品种间变异系数反映了性状在不同品种间的离散情况。由各性状变异系数计算结果(表 2)可知,18 个数量性状在品种内稳定性较好,花梗长度品种内变异系数最高,为 0.22,其余均未超过 0.2,均值为 0.13;花部性状的品种间变异系数较大,而叶部、茎部等性状在品种间的差异不如花部性状明显。因此,在关联分析中可能更容易在不同基因型间找到与花部性状关联的标记。
2.2 SRAP 分析在 130 对 SRAP 引物组合中筛选出 19 对条带清晰稳定的引物组合对所选择的 58 个大菊品种进行扩增,共产生扩增片段 283 条,其中多态性片段为 225条,多态性位点占 80%。每对引物组合检测等位基因数为 7—23 个,平均为 11.84 个(表 3)。多态性信息量 PIC 值在 0.76—0.94,平均为 0.87,为高度多态性座位,说明选择的大菊品种群体的遗传差异非常大,具有丰富的遗传多样性,这有利于在关联分析中发现与表型性状关联的位点。图 1 是引物 Me1/Em6 在供试品种中扩增产物的 6%聚丙烯酰胺凝胶电泳图。
2.3 供试品种的群体结构分析
利用 SRAP 标记,基于数学模型的群体结构分析表明,样本的等位变异频率特征类型数 K=5(即服从Hardy-Weinberg 平衡的亚群数目为 5)时,其模型的后验概率[lnP(D)]最大(图 2)。由此判断 58 个大菊品种群体可被分为 5 个亚群。依据上述推测的 K 值绘制供试品种群体结构图(图 3)。从群体结构图可以看出,58 个大菊品种中存在 5 个亚群。分析各亚群的生物学意义,发现菊花品种划分与瓣型和地域相关,可基本被识别为平瓣类、管瓣类、畸瓣类、桂瓣类和日本品种亚群。各亚群存在明显的差异,具有一定独立性,这说明供试群体存在着明显的群体结构。日本品种亚群独立性最强,而桂瓣类亚群与平瓣类、管瓣类、日本品种亚群基因间渗透性最高;管瓣类亚群与平瓣类亚群有一定渗透性。
为避免群体结构的存在通过影响位点 LD 进而影响关联分析的准确性,本研究将各品种 Q 值(各品种个体归入各亚群的概率)作为协变量纳入 SRAP 标记与表型性状变异的回归分析中。
2.4 SRAP 标记与表型性状的回归关联分析
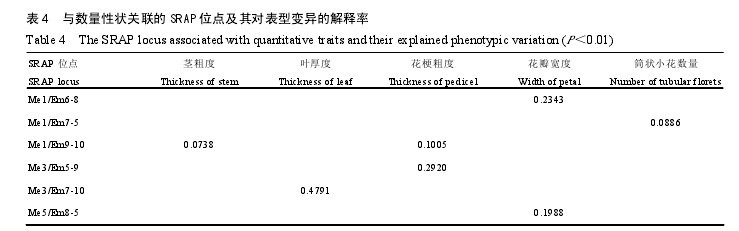
将58个大菊品种各品种相应的Q值作为协变量,将18个数量性状的表型变异对SRAP标记变异进行回归分析,寻找与性状相关联的标记及其等位变异。结果显示,在所检测的 225 个 SRAP 标记位点中,共有6 个 SRAP 标记位点与 5 个数量性状(茎粗度、叶厚度、花梗粗度、花瓣宽度和筒状小花数量)在 P<0.01水平上相关。与花部性状(花梗粗度、花瓣宽度和筒状小花数量)相关位点共 5 个,与茎部(茎粗度)、叶部性状(叶厚度)相关位点各 1 个,其中位点Me1/Em9-10 同时与茎粗度、花梗粗度相关。各位点对表型变异的解释率在 0.0738—0.4791,解释率最大(0.4791)的 SRAP 位点是 Me3/Em7-10,与叶厚度相关,而解释率最小(0.0738)的位点是 Me1/Em9-10,与茎粗度相关(表 4)。【表4】
3 讨论
3.1 基于数学模型的菊花群体结构分析
利用 SRAP 标记,使用 STRUCTURE 软件对供试菊花种质进行的基于数学模型的类群划分,与笔者基于遗传距离的 SRAP 标记 UPGMA 聚类结果(未列出)基本一致。发现中国大菊品种可分为平瓣类、管瓣类、畸瓣类和桂瓣类 4 个亚群结构,各亚群结构存在广泛的基因交流,匙瓣类品种并未单独形成亚群,而是分散在平瓣类和管瓣类亚群内,说明匙瓣类品种在演化关系中是处于平瓣类和管瓣类之间的过渡状态,这与张树林提出的不把匙瓣作为单独瓣型的观点相一致。尽管所选日本品种多为平瓣类和管瓣类,但并未与中国大菊的平瓣类与管瓣类聚为一群,而是单独聚为 1 个亚群,说明可能由于地理隔离的因素,其种质来源与演化过程与中国品种相比有一定的差异;但其与各亚群也都存在基因交流,表明日本品种和中国品种在菊花的栽培育种史上存在广泛的种质渗透现象。
本研究利用 SRAP 基于数学模型的菊花群体结构分析与前人利用形态学数据或其它分子标记基于遗传距离的聚类方法得到的结果基本一致,证明其可有效对菊花群体结构进行判断和划分。相比基于遗传距离的聚类方法,利用数学模型来分析群体结构可排除亚群划分的人为因素,更精确地确定亚群数目,并且可更清晰地观察各亚群间的基因交流情况。
3.2 与表型性状关联的分子标记位点
在植物中首次进行全基因组关联分析的是 Hansen等对野生甜菜生长习性的研究,发现 17 个引物组合扩增的 440 个全基因组范围内的 AFLP 标记中,有2 个与控制抽薹前是否需要春化的 B 基因显着关联。
Kraakman 等对236个AFLP标记和春大麦品种的产量及产量稳定性进行了关联分析,分别发现 8 个和 5个 AFLP 标记与产量和产量稳定性相关联。何静等应用SRAP和EST-SSR分子标记对木薯品种农艺性状进行关联分析,在 1471 个 SRAP 标记位点中共检测到73 个位点与 21 个农艺性状相关联,其中 46 个 SRAP标记位点同时与多个性状相关联;在 993 个 EST-SSR标记位点中有 20 个位点与 20 个性状变异相关。本研究在 225 个 SRAP 标记位点中共检测到 6 个位点与大菊品种 5 个数量性状相关(P<0.01),与数量性状相关联的 SRAP 位点较少,可能是由于 SRAP标记数量较少造成。严格意义上讲,全基因组关联分析需要成千上万的标记以及尽可能多的无亲缘关系的个体。即使是基因组很小的拟南芥,也需要检测 2 000个分子标记。本研究在 18 个数量性状中只找到 5个有标记与之关联的性状,除了标记数量较少外,供试群体的大小也是影响因素之一。本研究选择的菊花品种群体较小,每个瓣型的代表性品种约 10 个,每个花型的代表性品种约 2 个,因此在表型性状多样性分析中可能并未完全覆盖各个性状的全部变异,SRAP标记检测到的变异位点也十分有限。因此本研究只能算粗略意义上的全基因组关联分析,在进一步的关联分析研究中还需增加标记密度,增大群体规模。
本研究所找到的关联位点中,与花部性状显着相关 SRAP 位点共 5 个,而与茎部、叶部性状相关位点各 1 个,这可能与花部性状在品种间存在较大程度变异有关,更利于关联标记的发现。位点 Me1/Em9-10同时与茎粗度、花梗粗度相关。与花梗粗度相关的位点有 2 个,分别是 Me1/Em9-10 和 Me3/Em5-9,与花瓣宽度相关的位点有 2 个,分别是 Me1/Em6-8 和Me5/Em8-5。在中国大菊中,最为困扰育种者的难题之一就是由于花梗纤细造成花头易折,需要绑扎,耗费人力物力。本研究发现与花梗粗度相关联的位点 2个,这为大菊育种中培育粗壮品种提供了依据。
目前应用于植物关联分析的分子标记主要是 SSR和 SNP 标记,本研究应用的是 SRAP 分子标记。SRAP标记位点的多态性比 SSR 或 EST-SSR 高,并结合了RAPD 和 AFLP 的优点,引物的多态性可能由于包含了内含子、启动子和间隔序列在品种间的变异而产生,具有简便、稳定、高通量等特点。但是 SRAP 应用于关联分析也存在缺点:SRAP 标记是显性标记,不能区分纯合显性和杂合显性;不能直接获得位点大小信息,需要根据 marker 片段估测,增大了位点统计误差。因此在以后的菊花关联分析中增加 SSR(或EST-SSR)、SNP 等共显性分子标记是研究的趋势,而菊花中尚未开发出这两类标记。鉴于目前菊花基因组的研究水平,利用各种方法开发出菊花的 SSR 标记是目前迫切并且可行的研究任务。
3.3 菊花的关联分析与连锁作图
家系连锁作图(Family-based mapping,FBLmapping)与关联分析是解析植物重要数量性状基因型的主要方法。本研究利用关联分析的方法通过回归分析检测菊花品种表型变异和 SRAP 位点等位变异的关联性。在缺乏高密度菊花分子遗传连锁图谱的情况下,利用自然群体开展 QTL 分析,无疑是一种较简便且有效的方法。研究表明,利用关联分析检测到的分子标记位点,大多与定位于遗传连锁图上的 QTL 位点相一致。但关联分析的缺陷是不能估计 QTL 的具体位置及其加性或上位性效应。如果关联分析和连锁作图相结合,将大大促进复杂数量性状的剖析,有助于更准确、更精确地鉴定出控制菊花重要园艺性状的 QTL位点。
4 结论
本研究是首次在菊花品种中开展利用自然群体的关联分析。使用筛选出的 19 对 SRAP 引物组合对 58个典型大菊品种进行多位点扫描分析。在对供试材料进行基于数学模型的群体结构分析的基础上,将菊花18 个重要表型性状数据分别对 SRAP 标记变异进行回归分析。检测到 5 个数量性状与 6 个 SRAP 标记位点相关联,获得了与茎粗度、叶厚度、花梗粗度、花瓣宽度和筒状小花数量关联的标记位点。本研究证明基于数学模型的群体结构分析可有效对菊花群体结构进行判断和划分,也为大菊品种分类提供了新的手段;关联分析为菊花复杂的数量性状解析提供了新的途径,能够有效地找到与菊花表型性状关联的 SRAP 标记。研究结果可为菊花新品种选育、分子标记辅助育种和控制优异性状的相关基因的克隆奠定基础。
References
[1] 戴思兰. 中国菊花与世界花卉园艺. 河北科技师范学院学报, 2004,18(2): 1-7.Dai S L. Chinese florist’s chrysanthemum and the world flowerhorticulture. Journal of Hebei Normal University of Science ﹠Technology, 2004, 18(2): 1-7. (in Chinese)
[2] 张莉俊, 戴思兰. 菊花种质资源研究进展. 植物学报, 2009, 44(5):526-535.





