
基因定点敲除技术通过对染色体上基因特异位点进行定点突变,使其功能丧失,进而达到研究其功能的目的。一些利用基因敲除技术创建的动物疾病模型对医学研究的发展做出了重要贡献。传统基因敲除技术于上世纪 80 年代末由 Smithies 等[1]创立,通过同源重组手段将构建的外源 DNA 片段插入并替换相应染色体上特定的区段,达到基因敲除的目的。这一技术的应用使人类能够有目的地研究基因功能和与之相关的疾病之间的直接关系。然而由于同源重组技术需要载体构建、ES 细胞打靶、筛选、显微注射、小鼠鉴定等一系列较为复杂的操作步骤,并且成本较高、周期较长,大大限制了这一技术的应用。针对同源重组的缺点,科学家一直在探索用其他更简便的技术来实现基因敲除。先后发明了ENU 随机突变、基因捕获、ES 细胞基因敲除细胞库等方法和手段,但由于技术手段和操作成本的限制,这些技术都不能完全替代传统的同源重组技术。
2009 年以后出现了一系列新的基因编辑技术,包括ZFN、TALEN 和 CRISPR / Cas9,开创了基因敲除( 敲入) 新的时代。这些新基因编辑技术作用原理相似,即通过核酸酶在目的基因的 DNA 双链上制造切口,然后利用生物体自身的修复机制以同源重组或者非同源末端连接的方式实现修复,进而达到基因失活的目的。作为一种最新的基因编辑技术,CRISPR / Cas9 技术通过 gRNA 识别特异的靶序列,同时介导 Cas9 核酸酶在 DNA 的特异位点上制造切口[2],然后利用生物体自身的修复机制进行 DNA 修复。这一过程中可能造成碱基改变、增加、缺失等突变现象,进而实现对目的基因的敲除与修饰[3].
CRISPR / Cas9 系统的高效基因组编辑功能已被成功应用于多种生物。包括人类细胞,斑马鱼[5]、小鼠[6,7]、大鼠[8]、植物[9,10]及细菌[11].众所周知,生物体的一个性状往往由多个基因共同决定,传统的基因敲除费时费力却只能一次敲除一个相关基因,而 CRISPR/Cas9 系统其中一个最重要的特点便是可在多个不同 gRNA 的引导下由 Cas9 核酸酶一次靶向敲除多个基因组位点[12,13].与其他基因编辑技术相比,CRISPR/Cas9 系统具有其独特的优势: 首先,其 RNA 识别 DNA 机制即核酸之间的相互识别,避免了 DNA 甲基化造成的影响。其次,与ZFN 和 TALEN 相比,具有相同或更高的基因编辑效率,尤其在点突变方面优于 ZFN 和 TALEN[14,15].
第三,设计简单快速,无需重复构建核酸内切酶表达载体,适用于任何分子生物学实验室。
当前基因编辑技术迅速发展,CRISPR/Cas9 技术以其优势必将迅速推动基因组功能的研究。快速获取目标 gRNA 并检测验证其活性将会推动这项技术的发展。本文旨在建立一种体外 gRNA 快速合成及检测的方法,这一方法的建立将减少利用该系统实现基因编辑的时间,快速实现目标基因的敲除。
1 材料和方法
1. 1 材料
8 周龄 SPF 级 C57 小鼠,由北京华阜康生物科技股份有限公司提供【SCXK( 京) 2014-0004】,PCR仪( Bio-Rad T100TM Thermal Cycler) 、Tanon 全自动数码凝胶成像分析系统( 上海天能公司) 、NanoDrop2000 超微量分光光度计( Thermo) 、T7 转录试剂盒( Ambion_mMESSAGE_mMACHINE_T7) 、RNA 纯化试剂盒( MEGAclearTMKit,Ambion) 、PCR 产物纯化试剂盒( QIAquick PCR Purification Kit) 、无核酸酶水、dNTP mixture、高保真酶 Taq 酶( Sigma) 、Cas9 核酸酶( 北京唯尚立德生物科技有限公司)
1. 2 gRNA 靶点选择及其寡核苷酸链合成
利用设计能够结合NKp46 外显子 Exon3 上的 gRNA ( 20nt) .引物及gRNA 通用模板由 Invitrogen 公司合成。
1. 3 gRNA 转录模板的 PCR 扩增利用
NKp46 gRNA 特异的上游引物 mNKp46_g1F 和 gRNA 通用下游引物 gRNA-R,以人工合成的gRNA 通用模板 DNA 片段为模板,通过 PCR 扩增得到 NKp46 基因特异的 gRNA 转录模板。具体反应条件如下: ( 94℃ 30s,60℃ 10s,72℃ 30 s) × 35,72℃10 min,4℃ 保存。取 5 μL 电泳检测 PCR 产物,确定得到目标 DNA 片段,大小为 123 bp.
1. 4 gRNA 模板的体外转录
在得到 gRNA 转录模板后利用纯化试剂盒进行gRNA 转录模板的纯化。 将纯化后的产物用 T7RNAkit 转录试剂盒 ( Ambion _ mMESSAGE _ mMA-CHINE_ T7 ) 进行 NKp46 gRNA 体外转录并利用RNA 纯化试剂盒( MEGAclearTMkit,Ambion) 将转录产物进行纯化。合成的 gRNA 通过 NanoDrop 2000超微量分光光度计检测检测浓度及纯度。分装并 -80℃ 冻存。
1. 5 gRNA 体外活性及特异性检测
将得到的 gRNA 与 Cas9 核酸酶以及目的 DNA混合孵育,电泳后观察条带大小并观察片段灰度来确定切割效率。反应体系: dsDNA( 4μL) ,gRNA( 体外转录) ( 1μL) ,Cas9 核酸酶( 1μL) ,10 × buffer( 2μL) ,ddH2O( 12μL) ,反应总体系( 20μL) 按照上述反应体系,混合好反应溶液,37℃ 30 min,65℃5 min.
2 结果
2. 1 小鼠 NKP46 基因 gRNA 靶点选择
NKp46 基因存在于啮齿类动物 ( 大鼠和小鼠)和灵长类动物,如黑猩猩和短尾猿中。同时 NKp46是唯一能被小鼠细胞上表达配体所识别的人类 NK细胞激活受体。为了研究 NKP46 基因在病毒感染过程中的作用,我们希望通过 Cas9 技术创建 NKP46敲除小鼠。设计得到 gRNA,见表 1.
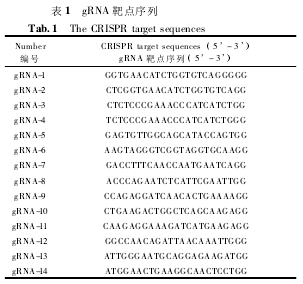
本文以小鼠 NKP46 基因为例设计 gRNA.共选择了 2 条 gRNA 序列:gRNA1 GGTGAACATCTGGTGTCAGG,gRNA2CTCGGTGAACATCTGGTGTC 这 两 个 gRNA 位 于Nkp49 基因组上外显子 Exon3 处。
2. 2 NKp46gRNA 体外扩增模板的设计
gRNA 转录模板的结构: T7 启动子、前导序列( 20nt) 、保守序列。体外扩增模板序列是固定的即gRNA 转录模板的保守序列 GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTT,该片段可以形成 gRNA 的发卡结构,是所有 gRNA 都包括的一部分。
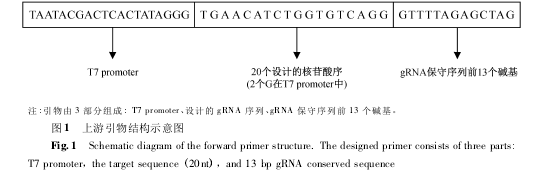
2. 3 上游引物 gRNA-F 的设计
将得到的 20nt 的序列前端加上一段 T7 启动子序列 TAATACGACTCACTATAGGG 并在末尾加上与gRNA 固 有 序 列 前 端 相 同 的 几 个 碱 基 GTTT-TAGAGCTAG,最终设计得到一个 53nt 的片段。按照该方法设计并合成了靶向小鼠 NKP46 基因的mNKp46_g1F.mNKp46 _g1F 将作为转录模板合成过程中的上游引物,参与 PCR 反应。由于体外转录试剂盒在转录完成后会在 5'端多出 3 个 G 所以在设计选择靶向 gRNA 时可以选择起始位置带 G 的序列,这样可以最大限度的减少体外转录的影响,它的构成 ( 图 1) mNKp46 _ g1F TAATACGACTCACTATAGGGTGAACATCTGGTGTCAGGGTTTTAGAGCTAG2. 4 gRNA 合成的通用下游引物。
gRNA-R 的设计 AAAAAAAGCACCGACTCGGT-GCCACTTTTTC,该序列与 gRNA 模板 3 '端互补。
2. 5 靶向小鼠 NKP46 基因的 gRNA 的转录模板在体外完成扩增
该过程十分简单只需一步 PCR 反应就可以迅速得到足够浓度的带有 T7 promoter 的 gRNA 的转录模板,参与反应的上游引物已经按照上述方法设计合成,其中下游引物和模板由于是通用的不具有特异性,不需要每次都合成。当研究者更换目标基因,只需设计合成一条新的上游引物即可,下游引物及模板不需要重复合成,十分的简单经济。扩增过程( 见图 2) .然后体外转录得到足够浓度 gRNA 的靶向 NKP46 基因的 gRNA.
2. 6 体外利用 Cas9 核酸酶检测验证该方法确实
可以得到高特异性及活性的 gRNA设计体外检测引物 mNk46_gSF TCCAAACACGTGCATGTTACACATG,mNk46 _ gSR CCCAGCTTTGACTCTCTTGTCCATC 以小鼠 DNA 作为模板,利用该引物可以扩增 NKP46 基因,作为体外酶切检测的片段。该片段中包含 gRNA 的靶点,利用这种体外检测方法,可以迅速对设计合成的 gRNA 的特异性及活性进行检测,通过电泳可以观察剪切条带的大小,以验证靶点位置是否正确,进一步可以通过 T-A 克隆检测切开位置。本文设计的靶向 NKP46 的 gRNA在靶点位置切开,使得全长 1297 bp 检测条带,被切成两条带,大小分别为 804 bp 和 493 bp( 图 3A) .
通过对灰度的计算( 图 3B) ,计算得到转录后原浓度的 gRNA 以及稀释一倍的 gRNA,剪切效率都为100% ,由此可以证明该方法设计合成 gRNA 特异性及活性较高,同时体外检测快速检测的方法大大提高了 gRNA 检测的效率。
3 讨论
常规获取 gRNA 要构建表达载体,需要把 gRNA的转录模板连接到载体上,经过涂板挑选单克隆再摇菌后提取质粒,最后还要酶切使质粒线性化才能得到 gRNA 的体外转录模板,得到 gRNA 最少也需要 3d 的时间。而本文建立的这种方法将极大地节约转录 gRNA 的时间,一步 PCR 反应就可以得到足够浓度的转录模板,仅需半天时间就可以得到需要的 gRNA.本文建立的这种活性及特异性检测方法,可以在体外迅速验证 gRNA 的其活性及特异性。
将 Cas9 核酸酶、gRNA、PCR 扩增的靶基因片段混合孵育,通过琼脂糖凝胶电泳可以十分清楚的观察到剪切位置并通过对电泳照片的灰度比较可以进一步计算剪切效率。
建立的这种快速利用 CRISPR /Cas 9 技术实现基因编辑的方法,最具特色的是 gRNA 的获得及体外酶切检测。与常规方法相比我们不构建 gRNA 表达载体而是直接利用化学合成方法。在体外合成一段单链 DNA,通过设计引物,利用 PCR 技术迅速扩增该片段,并转录纯化得到有功能的 gRNA.利用这种 gRNA 快速合成及检测的方法,可以使每次基因编辑仅需合成一段与目的基因互补的一小段DNA 链即一个上游引物即可,模板和下游引物都是通用的方便而经济。本文建立的体外酶切检测方法可以在得到 gRNA 后迅速对其活性及特异性检测,为后续转染或显微注射筛选有效的 gRNA 节省了宝贵的时间。





