
肝素(Heparin,HP)因其在肝中大量存在而得名,是一类长链糖胺聚糖,由硫酸化的葡萄糖胺和己糖醛酸以β-1,4糖苷键交叉连接组合而成.肝素在机体内能与多种蛋白质相互作用而具有多种生物功能,如抗凝血、抗炎和抑制肿瘤转移等.肝素分子量为3 000~50 000道尔顿,仅低分子的1/3部分有较强的抗凝作用.为此,近年国内外对低分子肝素的开发十分重视,希望通过化学降解或酶解的方法将肝素降解成平均分子量在4 000~6 500道尔顿的低分子量肝素(low molecular weight heparin,LMWH),由于其分子量较小,不易被血小板第Ⅳ因子中和,增强了抗凝效果和纤溶作用,同时抗血小板、诱发出血的副作用大大减少,几乎对脂质的代谢没有影响,因而倍受临床青睐.目前广泛用于心血管疾病和血液透析.
肝素酶(heparinases)是多糖裂解酶,作用于肝素或者硫酸乙酰肝素(heparan sulfate),可以将大分子肝素降解成低分子量肝素,由Payza等在肝素黄杆菌(Flavobacterium heparinum)中首次发现.此后,人们对肝素酶以及肝素酶生产菌作了广泛的研究.目前已从肝素黄杆菌中发现3种肝素酶:HepI、HepII和HepIII.LMWH的生产通常有化学裂解法和酶法.酶法生产LMWH具有低能耗、无污染、安全性高的优点,但也存在成本过高的缺点,主要是野生菌生产肝素酶的产量极低,而且需要价格昂贵的肝素诱导,由此导致其在生产上的使用受到较大的限制,因此如何获得廉价的肝素酶用于生产就成为酶法制备LMWH的关键.肝素酶I的异源重组表达是替代肝素黄杆菌生产肝素酶I的有效可行方案,对肝素酶基因进行克隆表达对解决这些问题并进一步探讨肝素酶的性质具有重要意义.
本文将HepI基因克隆到原核表达载体pGEX-4T-2中,并转化到大肠杆菌BL 21菌株中,通过IPTG诱导基因工程菌获得HepI酶蛋白,并对诱导与表达条件进行优化,为通过生物工程方法获得HepI酶蛋白奠定基础.
1 材料与方法
1.1 材料
肝素黄杆菌(F.heparinum),表达质粒pGEX-4T-2;大肠杆菌BL21菌株由本实验室保存;PfuTaq酶、dNTP、Sma I酶、Not I酶、T4DNA连接酶、蛋白质标准marker购自Ferments,DNA marker购自上海生工,其他试剂均为 国产分 析纯.肝 素酶HepI的PCR引 物1:5’AACCCGGGATGAAAAAA-CAAATTCTATA 3’(Sma I),引物2:5’AAGCGGCCGCCTATCTGGCAGTTTCGCTGT 3’(Not I)由生工上海有限公司合成.
1.2 方法
1.2.1 HepI基因的克隆取20μL肝素黄杆菌培养液到Ep管中,99℃裂解10min,10,000×g离心5min,取上清液1.0μL作为PCR扩增的模板,同时在PCR管中加入PfuTaq酶0.2~0.3μL,Buffer2μL,dNTPs 1.0μL,引物1和 引物2各1.0μL,H2O 14μL,反应总体积20μL,PCR扩增条件为:先94℃预变性3min,然后94℃变性30s,58℃退火30s,72℃延伸70s,共38个循环,最后72℃继续延伸10min.
1.2.2 HepI基因的重组将PCR产物经琼脂糖凝胶电泳检测后,切胶回收目的基因.取目的基因DNA约1.0μg用Sma I,Not I酶切,取pGEX-4T-2质粒DNA约1.0μg用Sma I,Not I酶切,然后回收酶切产物,取酶切过的目的基因DNA 200ng和质粒DNA 80ng在24 ℃下用T4DNA连接酶连接30min,用连接产物转化大肠杆菌BL21感受态细胞,涂布到LB固体培养基平板上(含氨苄青霉素)培养16h,挑取10个菌落,通过菌落PCR方法挑选阳性克隆,PCR条件同上.选取阳性克隆培养5mL菌液,提取质粒,通过双酶切法进一步检测,阳性菌落送生工测序.
1.2.3 HepI基因的诱导表达将测序正确的菌株接种到5mL含氨苄青霉素的LB液体培养基中,37℃下振荡培养过夜,第二天按1∶50的比例接种到新鲜的培养基中培养至生长对数期(约4h)后取出,放至室温后加入诱导剂异丙基-β-D-硫代半苷(IPTG),诱导剂的终浓度为0.25mmol/L的,然后将不同的菌液分别在不同温度下继续摇培4~6h进行目基因的诱导表达.离心法收集菌体,菌体用冰冷的PBS重悬,超声破碎裂解细菌,10 000×g离心10min,对离心后的上清和沉淀进行SDS-PAGE电泳分析.
1.2.4 HepI重组蛋白的纯化将含重组质粒的BL21菌按1∶50的比例接种于500mL LB液体培养基,摇培4h后取出冰浴10min,然后加入IPTG,在12℃继续摇培10h.离心法收集细菌,加入预冷的15mL 1×PBS缓冲液悬浮细菌,冰上超声裂解细菌.裂解液4℃下10 000×g离心20min,将离心后的上清液与Glutathione Sepharose 4B结合30min,将结合液装到层析柱上,用1×PBS洗去杂蛋白,然后用洗脱缓冲液(50mmol/L Tris-HCl,10mmol/L GSH,pH 8.0)洗脱目的蛋白.通过SDS-PAGE法鉴定融合蛋白的纯度,用Bradford法测定蛋白质浓度.
2 结果与分析
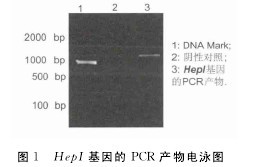
2.1 HepI基因的克隆与表达从NCBI获得HepI基因的DNA序列,以肝素黄杆菌的DNA为模板,用HepI基因的特异性引物扩增目的基因,扩增产物通过1.2%的琼脂糖凝胶电泳检测,可以在凝胶上看到1 000bp左右的条带,与预想的PCR产物大小相符合(图1).PCR产物经常规的操作步骤,构建出重组质粒pGEX-4T-2-HepI,然后转化大肠杆菌E.coli BL21感受态细胞,菌落PCR扩增出1 000bp大小的特异性条带.【图1】
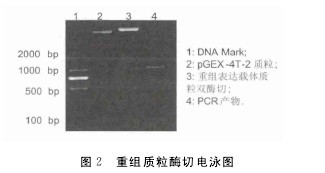
对该重组表达质粒进行双酶切鉴定,也得到一条长约1000bp的条带,与预期大小一致(图2),说明HepI基因已克隆到表达载体上,对酶切鉴定正确的阳性克隆进行测序,使用DNAMAN软件比较此次克隆到的序列和NCBI上的序列,结果表明序列正确,没有移码或突变,表明原核表达载体成功构建.重组菌用IPTG诱导,SDS-PAGE电泳结果表明,经IPTG诱导后,重组菌出现一条非常明显的诱导蛋白条带,条带大小在58U(图3),此带的大小与肝素酶HepI蛋白(约30U)及与其融合的GST蛋白的分子量(28U)之和大致相当.【图2-3】

但在对照组(未经IPTG诱导的重组菌株)中看不到该蛋白条带,说明HepI基因插入表达载体pGEX-4T-2后,在大肠杆菌中获得了高效表达,但在37 ℃下诱导形成不可溶的包涵体.改变诱导温度,经过不同温度的多次尝试,发现融合蛋白在12℃条件下部分可溶.
通过诱导温度的试验,我们选择12℃作为肝素酶的诱导温度,重组菌经IPTG诱导12h后离心收集菌体,超声波破碎,Glutathione Sepharose 4B亲和层析纯化.
纯化的重组蛋白进行SDS-PAGE分析以鉴定其纯度,结果在凝胶上只有一条58U大小的条带,说明获得了较高纯度的重组融合蛋白(图4).【图4】

3 结论
通过本研究,我们获得了表达肝素酶HepI的基因工程菌,并对肝素酶的表达条件进行了初步优化,采用亲和层析方法,获得了重组表达的融合酶蛋白,为肝素酶的应用研究打下了较好的基础,同时为开展重组酶蛋白降解肝素的活性与降解条件研究创造了条件.





