摘要:生物大分子的微观结构动力学决定其生物学功能, 其力学-化学耦合规律是分子生物力学的重点关注方向。分子动力学模拟是耦合生物大分子力学-化学性质微观结构动力学基础的有效手段, 其结果可用于预测结构-功能关系、指导实验设计和诠释实验结果。本文简要介绍了分子动力学模拟的方法学特点、基本工作原理及其在分子生物力学中的应用, 并展望了未来可能的发展方向和应用前景。
关键词:分子动力学模拟; 分子生物力学; 微观结构动力学; 结构-功能关系;

Molecular Dynamics Simulation and Molecular Biomechanics
Abstract:
Micro-structural dynamics of biomolecules governs their biological functions.Mechano-chemical coupling is a key issue in molecular biomechanics.Molecular dynamics simulation (MDS) is an effective approach to coordinate the biomolecular micro-structural dynamics with their mechanical and chemical features.The outcomes provide the bases in predicting the structure-function relationship, optimizing the experimental design, and interpreting the measured data.This mini-review briefly introduces the MDS approach, the working principle, and the biological significance in molecular biomechanics, and proposes the prospects of future development and potential applications.
Keyword:
Molecular dynamics simulation;Molecular biomechanics;Micro-structural dynamics;Structure-function relationship;
引言
生物大分子是生物体生命活动的直接执行者。比如, 细胞粘附分子及其配体间的特异性相互作用, 是介导细胞聚集和粘附动力学行为、实现细胞功能的必要媒介;信号蛋白分子是感受、传递并转导细胞信号的桥梁;通道蛋白分子是维持细胞内外小分子物质及其离子平衡的决定因素等。而生物大分子的微观结构特征及其动力学信息, 是实现、调控其生物学功能的基础和关键[1,2]。例如, 受体-配体间的特异性识别需要结构上特异性的匹配;信号分子不同结构态之间的变构是将上游信号传递给下游, 从而启动信号传递和转导的重要途径;不同通道蛋白的结构特征是特异性识别不同通透物质、调控通透门控机制的基础等。因此, 考察生物大分子的微观结构动力学特征, 是理解其结构-功能关系的重要研究内容。
X-射线晶体衍射、核磁共振波谱和电镜三维重构是研究生物大分子结构的三种主要实验方法。其中, 核磁共振波谱技术是能够在原子分辨率下测定溶液中生物大分子三维、动态结构的唯一方法, 但目前还受到待测蛋白分子量的限制, 仅能测量分子量相对较小的分子[3]。电镜三维重构是一种新型的结构生物学研究方法, 一方面, 其解析生物大分子复合体结构的分辨率日益提高, 可以提供大蛋白分子或复合体的精细结构;另一方面, 还可以解析生物大分子在不同功能状态下的结构及变化, 对于揭示生物大分子复合体结构的作用机理具有重要作用[4,5];而且, 该方法比较适合研究分子量巨大、组成复杂、具有结构多样性的生物大分子复合体。但相对其他两种手段来讲, 电镜三维重构技术目前的分辨率还较低, 通常得到的是生物大分子的外在轮廓或主链信息。X-射线晶体衍射是比较经典的结构生物学研究方法, 精度较高, 而且适用对象范围较宽, 但其缺点是尚无法得到动态结构信息, 而且, 大分子可否结晶以及结晶的好坏也是限制该技术的瓶颈之一。综上所述, 结构生物学主要研究方法的发展为诠释生物大分子功能的结构基础提供了强有力的技术支持, 大大推动了生物大分子结构-功能关系的研究。但基于各自的技术特点, 目前还没有可以提供动态的高精度结构信息的有效实验手段, 而且, 由于实验设备、实验技术、实验样品准备、实验周期及实验成本等的限制, 难以在普通的生物学实验室得到普及。随着计算机技术的发展, 计算科学在科学研究中占据越来越重要的地位, 而分子动力学模拟 (molecular dynamics simulation, MDS) 作为计算科学的重要研究手段之一, 以其可以同时提供高空间分辨率微观结构信息及高时间分辨率结构动力学信息的独特优势, 在生物大分子微观结构动力学研究中发挥着重要作用[6~9]。目前, MDS与实验手段相结合, 在生物大分子结构-功能关系研究中得到越来越广泛的应用。
生物力学是结合生物学与力学的原理和方法, 研究生命体运动和变形的学科。其研究内容包括器官、组织、细胞、分子、基因等具有不同时间和空间特征尺度的多层次耦联行为。分子生物力学是生物力学研究中的重要组成部分, 其关注重点是定量研究生物大分子的力学-化学耦合规律, 主要研究内容包括定量描述生物大分子间特异性相互作用动力学、蛋白质组装动力学、作用力对蛋白质间相互作用的调控等。随着分子生物力学研究技术与方法 (如微管吸吮术、原子力显微术、光镊、生物膜力探针和平行流动腔等) 的发展, 以及其理论模型[如小系统概率动力学模型、一阶不可逆反应动力学、动力学力谱 (dynamic force spectroscopy, DFS) 等]的不断完善, 分子生物力学的研究不断深入[10]。基于此, 在炎症级联反应、肿瘤转移等病理生理过程中起重要作用的选择素-配体、整合素-配体反应动力学及其外力调控规律得到了很好的定量描述[11~16], 并先后发现了与物理直观相悖的“逆锁键”行为[17~19]。已有研究表明, 不同分子体系具有各自独特的反应动力学特性, 而且对外界物理因素 (比如外力) 的响应规律也存在差别;为研究分子体系的内在机理, 同样需要其微观结构动力学信息, 以解释其定量生物力学规律的差异, 从而更好地理解其生物学功能。因此, 分子动力学模拟同样是分子生物力学研究中考察微观结构动力学基础的重要研究手段。
结构决定功能。考察目标分子的微观结构动力学是理解其生物学功能的基础与根本, 同时也是进一步有针对性地调控其功能的前提。分子动力学模拟方法作为研究生物大分子微观结构动力学的有效手段, 在生物大分子的结构-功能关系研究中具有举足轻重的地位, 并有广阔的发展空间。
分子动力学模拟方法简介
分子动力学模拟方法的基本原理是, 以原子为基本元素, 以牛顿第二定律为控制方程, 在经验势场作用下, 采用计算机模拟由多个原子组成的分子体系随时间变化的动态演化过程。其基本工作流程如图1所示:首先是模拟系统的构建, 主要包括目标分子初始原子坐标和模拟系统环境构建两部分。前者主要通过蛋白质数据库 (Protein Data Bank, PDB) 获得, 对于蛋白质数据库中尚未报道的结构, 也可通过同源模建、分子对接等模拟手段获得其初始结构;后者则根据目标分子的生物学特征构建相应水环境、离子环境和质膜环境等, 以期尽可能模拟生理环境。能量最小化是优化模拟系统的重要过程, 共轭梯度和最陡下降算法是能量最小化的常用算法, 其目的是降低系统构建过程中产生的不合理构象, 并且使系统能量达到极小或最小;然后经过系统升温, 进行分子动力学模拟的主要模拟过程———平衡模拟, 该过程是考察目标分子微观结构特征及其结构稳定性的主要模拟过程, 同时也是进行后续调控模拟的必要过程。目前, 分子动力学模拟的方法学平台较多, 而且日趋成熟。广泛用于生物大分子结构动力学研究的主要程序包括:CHARMM[20]、Amber[21]、NAMD[22]、GROMACS[23]和ACEMD[24]等。不同程序虽具有类似的系统建立、动态模拟等运行流程, 但各自处理问题的着重点稍有差别, 需要研究者根据模拟目标来进行选择。势场参数是描述模拟体系中任何两个原子之间相互作用的参数, 既包含原子之间相对空间定位的描述, 又包含原子之间相互作用强弱的定义, 是分子动力学模拟的必要条件[25]。原子间的相对空间定位如图2所示, 可分为4类。基于该空间定位, 生物大分子系统研究中应用比较广泛的经验势场大致分为两类:ClassⅠ和ClassⅡ[26]。ClassⅠ的一般表达式为:
图1 分子动力学模拟基本流程Fig.1 Flowchart of molecular dynamics simulation

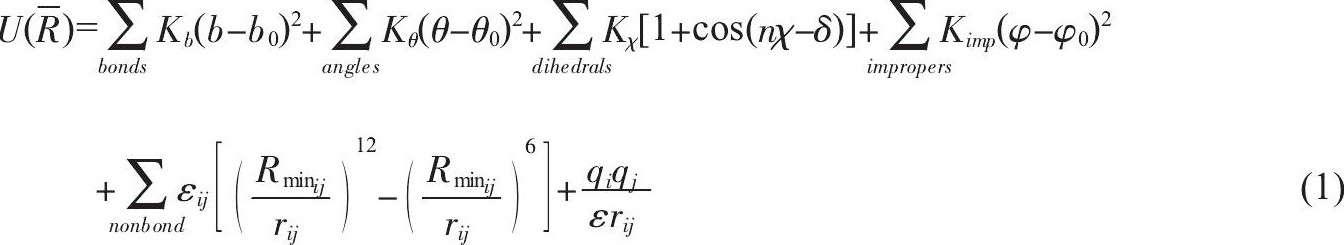
其中, 包含了描述生物大分子系统相互作用的最基本作用项。等号右边的前四项属于共价相互作用, 包括两原子间共价相互作用 (bond) 、键角相互作用 (angle) 、二面角相互作用 (dihedral) 和非共面扭转作用 (improper) ;后两项是描述非共价相互作用的范德华相互作用和静电相互作用。其中, b为键长、θ为键角、χ为二面角、准为非共面扭转角。相应的平衡几何参数和力常数分别为:平衡键长b0和力常数Kb, 平衡键角θ0和力常数Kθ, 二面角周期n、相角δ和力常数Kχ, 非共面平衡扭转角准?0和力常数Kimp, 原子i与j间的最小作用半径Rminij及对应最小作用半径的势垒高度eij, e为介电常数。
图2 原子间共价相互作用模式图Fig.2 Covalent interaction of adjacent atoms

根据研究目的, 可选择性地进行调控模拟过程, 调控模拟是考察外界因素调控规律的模拟过程, 该过程一方面可以在模拟能力允许的范围内加速实现特定生物学过程, 另一方面, 也是特定分子体系具有生物学意义的调控模拟过程。为了实现特定调控过程, 对应的模拟方法学也相应出现, 逐渐发展成为模型化程序。比如, 为了考察生物体内受到外力调控的一些蛋白的 (如微丝蛋白Actin) 力学性质, 以及细胞粘附分子与配体间相互作用 (如选择素-配体、整合素-配体相互作用) 的力学调控规律, 调控分子动力学 (steered molecular dynamics, SMD) 出现, 并被广泛应用[27]。该方法最早是NAMD模拟程序中的一个功能模块。受原子力显微镜技术中力学加载模式的启发, SMD可以进行恒速或恒力加载, 目前该思想已经被广泛扩展并在多种模拟程序中应用。生物大分子的变构动力学与其功能直接相关, 整合素的不同构象态表征其不同的激活程度, 同时调控不同的细胞粘附动力学行为。信号分子的变构行为是实施其信号感知、传递和转导的途径。因此, 考察生物大分子变构特征及其动力学过程是非常重要的研究内容。但由于模拟能力限制, 难以在模拟时间内观察到变构过程, 基于此, 目标分子动力学 (targeted molecular dynamics, TMD) 出现, 并在多种分子体系变构模拟中得到应用[22]。TMD同样是NAMD中的一个功能模块, 其工作原理是根据感兴趣分子现有构象和目标构象之间的差别, 在模拟时间内逐步施加外力至每个原子上, 使得分子构象向目标构象靠近。虽然该过程是人为调控而不是自然发生的真实过程, 但通过考察其动力学过程可预示变构发生的可能路径。另外, 血液剪切流场是生物体内许多蛋白分子所处的力学微环境, 并会对其构象动力学产生影响, 为准确评估目标分子微观动力学特征, 需要建立与生理环境更加接近的流动剪切体系。基于此, 流动剪切分子动力学[ (shear) flow molecular dynamics, (S) FMD]的研究也相继出现[28~30], 其基本原理是通过在模拟体系的水分子上施加外力, 使其产生定向运动, 从而形成微观流场。但是, 与SMD和TMD相比, (S) FMD的方法学还有待进一步完善并程序化。总之, 调控模拟是范围宽广的模拟过程, 调控模式会根据模拟目标而变化。随着分子动力学模拟方法在生物大分子结构-功能关系研究中的进一步应用, 调控模拟模式会越来越多样化, 一些常规模式也会越来越程序化。对模拟结果的分析、总结与完善, 以及根据实验研究进行的迭代过程, 是回答生物大分子力学-化学耦合问题的关键, 虽然与模拟不直接相关, 但同样是分子动力学模拟技术中必不可少的环节, 模拟结果能否为现有实验结果提供微观结构解释、或者为实验提供预测信息并指导后续实验设计, 都依赖于对模拟数据的分析、挖掘和提炼。但因为模拟对象的多样化, 难以规定固定的数据分析模式, 需要根据研究对象和内容进行分析。
分子动力学模拟在生物大分子研究中的应用
自1977年首次成功运用分子动力学模拟研究蛋白质性质以来[31], 该方法已被广泛应用于生物大分子微观结构动力学的研究, 大概可以分为以下几类:一是单分子的折叠及去折叠模拟。比如蛋白质L与G的折叠过程模拟[32], 蛋白分子Titin和Fibronectin[33,34]、P-选择素[35]和v WF的去折叠模拟[36], 以及DNA的去折叠过程模拟[37]等, 该类研究为考察生物大分子内部不同结构域的微观结构特征及其稳定性提供了基础。二是外力作用下分子复合物的解离模拟。比如生物素-亲和素[38]、CD2-CD58[39]和P-选择素-PSGL-1[40,41]等复合物的解离模拟, 该类模拟为研究受体-配体特异性相互作用的微观结构基础、相互作用强弱, 以及外力调控相互作用规律等提供了线索。三是生物大分子的变构模拟。比如DNA变构动力学模拟[37], 以及β2整合素[42,43]、Glycoprotein Iba[44]、P-选择素[45]等的变构模拟, 该模拟可为预测生物大分子的变构途径及其不同构象态与功能的关联等提供信息。目前, 分子动力学模拟在生物大分子结构动力学研究中的应用日趋广泛, 研究对象多种多样 (已从可溶性蛋白向膜蛋白扩展[46]) , 研究内容也千差万别 (包括水通道蛋白水通透性能力模拟[47,48]、钾离子通道蛋白门控机制模拟[49~51]、荧光膜探针分子与质膜相互作用模拟[52]、环境调控的蛋白质-蛋白质相互作用及其聚集过程模拟[53]等) 。总之, 分子动力学模拟在生物大分子结构-功能关系研究中的广泛应用, 大大提高了人们对其生物学功能微观结构动力学机理的认识, 拓展了分子生物力学研究的深度。
分子动力学模拟方法学的发展
分子动力学模拟的广泛应用离不开方法学的快速发展和完善。生物大分子是相对复杂的模拟体系, 体现在生物学环境的复杂性, 以及分子自身空间尺度构象和时间尺度动力学的复杂性。尽可能模拟目标分子的真正生物学环境及动力学行为与计算能力是相互矛盾的两个方面, 研究者需要根据自己的研究目标平衡对模拟精度和速度的要求。自分子动力学模拟出现至今, 研究者们也一直在这两个方面不懈努力, 并已取得较大进展。在追求模拟精度方面, 一方面是发展新的方法学以尽可能模拟生理环境和调控, 如分子动力学与量子力学的结合将非共价相互作用模拟拓展至共价相互作用模拟[54,55];剪切流场分子动力学及膜环境的建立等, 均为更加真实地模拟生理环境提供了基础。该方面研究以增加模拟体系复杂性为代价。另一方面则是完善模拟算法, 增强模拟采样有效性, 在不过多增加模拟体系复杂性的基础上, 提高“信噪比”。加速分子动力学 (accelerated molecular dynamics, AMD) 是典型代表之一[56~58], 该方法是偏置势能分子动力学的拓展, 可以在事先不知道自由能表面特征的条件下, 有效加强构象空间的遍历, 而且不需要事先定义反应路径或特定采集变量。在追求模拟速度方面, 除了发展分子动力学模拟的自身算法[59], 也可从模拟方法学上进一步改进。一方面, 粗粒化模拟方法是典型代表[60,61], 其代价是降低精细结构信息;另一方面, 可通过外界调控加速模拟过程, 比如SMD或TMD, 虽然有模拟生理调控的考虑, 也有加速模拟过程的考虑, 其代价是会在一定程度上高估结构动力学的演化过程。另外, 分子动力学模拟的数据分析也受到研究者们的广泛重视, 除基于不同研究目的的相应分析外, 一些公用分析方法也发展迅速, 比如对目标分子亚稳态和变构动力学的分析[62], 以及对平衡模拟平衡判据的进一步完善[63]等。分子动力学模拟方法学和计算算法的发展和完善, 以及模拟数据分析和提炼方法的发展, 均为该技术平台在生物大分子结构-功能关系研究中的进一步推广打下了坚实基础。
总结与展望
生物大分子微观结构动力学特征决定了其生物学功能, 分子动力学模拟以其独特优势, 在生物大分子结构-功能研究中占据不可替代的位置。运用分子动力学模拟方法考察目标分子的结构动力学特征, 一方面可以从微观结构层次解释实验数据, 另一方面也可以预测、指导实验, 进一步改造分子功能。模拟与实验相辅相成, 互为补充。本文简要介绍了分子动力学模拟的必要性及基本原理和应用, 因为方法学的发展和应用涉及的领域非常广泛, 难以概括全面, 仅就我们课题组近年来在该方面研究工作中的理解进行归纳和整理, 希望对读者有所帮助。计算科学的快速发展、人们对生物大分子功能重要性认识的深入、以及结构生物学实验技术的推广, 均为分子动力学模拟方法学的进一步发展和完善, 及其在生物大分子结构-功能关系研究中的应用提供了良好的氛围。
技术的发展源于科学问题的驱动, 研究者们对生物大分子生物学功能微观结构基础的兴趣, 必将推动分子动力学模拟的发展及其在该领域的应用。与方法学发展相对应, 该方面今后的发展方向可简单归结为三个方面:一是模拟体系和模拟过程尽可能接近真实生物学环境, 包括目标分子自身完整性、目标分子真实生理环境模拟, 以及目标分子真实调控因素模拟等。目前, 基于各种条件限制, 多数模拟做了很大程度的简化, 比如仅考虑分子中的特定结构域、简化调控因素模式等。二是模拟算法的发展。目前, 模拟速度仍然是主要限制因素之一, 而模拟算法是提高模拟能力的主要途径之一。三是分析方法的进一步开发。生物大分子种类繁多, 功能千差万别, 分子动力学模拟信息量大, 如何从海量数据中提取有用信息, 同样是其服务于实验研究所面临的重要问题。
目前, 预测、指导实验及解释实验结果是分子动力学模拟在生物大分子结构-功能研究中的主要应用, 因此, 实验和模拟的直接对照是未来模拟追求的方向之一[64]。但因为计算能力和方法学的限制, 直接对照尚存在困难, 取决于实验测量的时间和空间尺度是否可以与模拟分析量的时间和空间尺度可比, 在二者之间架设桥梁是解决途径之一。目前, 基于反应路径一致的假设, 自由能计算是联系实验和模拟的纽带之一, 并取得了一定的进展。但自由能计算方法学还有待进一步完善[65]。
分子动力学模拟不仅在生物大分子结构-功能关系的基础研究方面起到重要作用, 同时也具有广阔的应用前景, 比如药物设计和药物发现[66]等。分子动力学模拟提供的生物大分子结构动力学及受体-配体相互作用的微观结构信息, 为药物设计和发现提供了重要线索。因为药物识别靶标分子并与其结合的过程是结构相互匹配的动态过程, 所以, 不同药物候选物与靶标分子相互作用动力学是评估候选物有效性的基础。药物开发是耗资巨大且需要大量人力、物力投入的漫长过程, 分子动力学模拟方法学的前期介入将大大减少实验的盲目性, 提高候选物的筛选效率, 从而起到节约人力、物力、财力, 缩短开发周期的目的。
参考文献
[1].Smock RG, Gierasch LM.Sending signals dynamically.Science, 2009, 324:198~203
[2].Tokuriki N, Tawfik DS.Protein dynamism and evolvability.Science, 2009, 324:203~207
[3].邓乾春, 黄庆德, 黄凤洪, 谢笔钧.蛋白质溶液构象的研究方法.生物物理学报, 2009, 25 (4) :237~246 Deng QC, Huang QD, Huang FH, Xie BJ.Progress of research method on protein solution conformation.Acta Biophys Sin, 2009, 25 (4) :237~246
[4].蔡刚.单颗粒电子显微学的研究进展.生物物理学报, 2010, 26 (7) :560~569 Cai G.State-of-the-art of single particle EM.Acta Biophys Sin, 2010, 26 (7) :560~569
[5].黄晓星, 宋晓伟, 朱平.冷冻电子断层成像技术及其在生物研究领域的应用.生物物理学报, 2010, 26 (7) :570~578 Huang XX, Song XW, Zhu P.Cryo electron tomography and its application to biological research.Acta Biophys Sin, 2010, 26 (7) :570~578
[6].Karplus M, McCammon JA.Molecular dynamics simulations of biomolecules.Nat Struct Biol, 2002, 9:646~665
[7].Mulholland AJ.Introduction.Biomolecular simulation.J R Soc Interface, 2008, 3:S169~172
[8].Van der Kamp MW, Shaw KE, Woods CJ, Mulholland AJ.Biomolecular simulation and modelling:Status, progress and prospects.J R Soc Interface, 2008, 3:S173~190
[9].Karplus M, Kuriyan J.Molecular dynamics and protein function.Proc Natl Acad Sci USA, 2005, 102:6679~6685
[10].Long M, LüSQ, Sun GY.Kinetics of receptor-ligand interactions in immune responses.Cell Mol Immunol, 2006, 3:79~86
[11].Huang J, Chen J, Chesla SE, Yago T, Mehta P, McEver RP, Zhu C, Long M.Quantifying the effects of molecular orientation and length on two-dimensional receptor-ligand binding kinetics.J Biol Chem, 2004, 279:44915~44923
[12].Wu L, Xiao BT, Jia XL, Zhang Y, LüSQ, Chen J, Long M.Impact of carrier stiffness and microtopology on two-dimensional kinetics of P-selectin and P-selectin glycoprotein ligand-1 (PSGL-1) interactions.J Biol Chem, 2007, 282:9846~9854
[13].LüSQ, Ye ZY, Zhu C, Long M.Quantifying the effects of contact duration, loading rate, and approach velocity on P-selectin-PSGL-1 interactions using AFM.Polymer, 2006, 47:2539~2547
[14].Zhang Y, Sun GY, LüSQ, Li N, Long M.Low spring constant regulates P-selectin-PSGL-1 bond rupture.Biophys J, 2008, 95:5439~5448
[15].Sun GY, Zhang Y, Huo B, Long M.Surface-bound selectin-ligand binding is regulated by carrier diffusion.Eur Biophys J, 2009, 38:701~711
[16].Fu CL, Tong CF, Wang ML, Gao YX, Zhang Y, LüSQ, Liang SL, Dong C, Long M.Determining beta2-integrin and intercellular adhesion molecule 1 binding kinetics in tumor cell adhesion to leukocytes and endothelial cells by agas-driven micropipette assay.J Biol Chem, 2011, 286:34777~34787
[17].Marshall BT, Long M, Piper JW, Yago Y, McEver RP, Zhu C.Direct observation of catch bonds involving cell-adhesion molecules.Nature, 2003, 423:190~193
[18].Sarangapani KK, Yago T, Klopocki AG, Lawrence MB, Fieger CB, Rosen SD, McEver RP, Zhu C.Low force decelerates L-selectin dissociation from P-selectin glycoprotein ligand-1 and endoglycan.J Biol Chem, 2004, 279:2291~2298
[19].Kong F, Garcia AJ, Mould AP, Humphries MJ, Zhu C.Demonstration of catch bonds between an integrin and its ligand.J Cell Biol, 2009, 185:1275~1284
[20].Brooks BR, Brooks CL, Mackerell Jr AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M.CHARMM:The biomolecular simulation program.J Comput Chem, 2009, 30:1545~1614
[21].Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz Jr KM, Onufriev A, Simmerling C, Wang B, Woods RJ.The Amber biomolecular simulation programs.J Comput Chem, 2005, 26:1668~1688
[22].Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K.Scalable molecular dynamics with NAMD.J Comput Chem, 2005, 26:1781~1802
[23].Hess B, Kutzner C, Van der Spoel D, Lindahl E.GROMACS 4:Algorithms for highly efficient, load-balanced, and scalable molecular simulation.J Chem Theory Comput, 2008, 4:435~447
[24].Harvey MJ, Giupponi G, De Fabritiis G.ACEMD:Accelerating biomolecular dynamics in the microsecond time scale.J Chem Theory Comput, 2009, 5:1632~1639
[25].González MA.Force fields and molecular dynamics simulations.Collection SFN, 2011, 12:169~200
[26].Mackerell Jr AD.Empirical force fields for biological macromolecules:Overview and issues.J Comput Chem, 2004, 25:1584~1604
[27].Isralewitz B, Gao M, Schulten K.Steered molecular dynamics and mechanical functions of proteins.Curr Opin Struct Biol, 2001, 11:224~23
[28].Lou J, Zhu C.Flow induces loop-to-beta-hairpin transition on the beta-switch of platelet glycoprotein Ib alpha.Proc Natl Acad Sci USA, 2008, 105:13847~13852
[29].Zou X, Liu Y, Chen Z, Cardenas-Jiron GI, Schulten K.Flow-induced beta-hairpin folding of the glycoprotein Ib alpha beta-switch.Biophys J, 2010, 99:1182~1191
[30].Kang YY, LüSQ, Ren P, Huo B, Long M.Molecular dynamics simulation of shear-and stretch-induceddissociation of P-selectin/PSGL-1 complex.Biophys J, 2012, 102 (1) :112~120
[31].McCammon JA, Gelin BR, Karplus M.Dynamics of folded proteins.Nature, 1977, 267:585~590
[32].Brown S, Head-Gordon T.Intermediates and the folding of proteins L and G.Protein Sci, 2004, 13:958~970
[33].Gao M, Craig D, Lequin O, Campbell ID, Vogel V, Schulten K.Structure and functional significance of mechanically unfolded fibronectin type III intermediates.Proc Natl Acad Sci USA, 2003, 100:14784~14789
[34].Gao M, Lu H, Schulten K.Unfolding of titin domains studied by molecular dynamics simulations.J Muscle Res Cell M, 2002, 23:513~521
[35].LüSQ, Long M.Forced extension of P-selectin construct using steered molecular dynamics.Chin Sci Bulle, 2004, 49:10~17
[36].Chen W, Lou JZ, Zhu C.Molecular dynamics simulated unfolding of von Willebrand Factor A domains by force.Cell Mol Bioeng, 2009, 2:75~86
[37].Perez A, Luque FJ, Orozco M.Frontiers in molecular dynamics simulations of DNA.Acc Chem Res, 2011, DOI:10.1021/ar2001217
[38].Izrailev S, Stepaniants S, Balsera M, Oono Y, Schulten K.Molecular dynamics study of unbinding of the avidin-biotin complex.Biophys J, 1997, 72:1568~1581
[39].Bayas MV, Schulten K, Leckband D.Forced detachment of the CD2-CD58 complex.Biophys J, 2003, 84:2223~2233
[40].LüSQ, Long M.Forced dissociation of selectin-ligand complexes using steered molecular dynamics simulation.Mol Cell Biomech, 2005, 2:161~177
[41].Lou JZ, Zhu C.A structure-based sliding-rebinding mechanism for catch bonds.Biophys J, 2007, 92:1471~1485
[42].Chen W, Lou JZ, Hsin J, Schulten K, Harvey SC, Zhu C.Molecular dynamics simulations of forced unbending of integrin alpha (V) beta (3) .PloS Comput Biol, 2011, 7:e1001086
[43].Mao DB, LüSQ, Li N, Zhang Y, Long M.Conformational stability analyses of alpha subunit I domain of LFA-1 and Mac-1.PLoS One, 2011, 6:e24188
[44].Huang Q, Lou JZ, Wu JH, Zhu C.Conformational transition of glycoprotein Iba mutants in flow molecular dynamics simulation.Cell Mol Bioeng, 2011, 4:495~504
[45].LüSQ, Zhang Y, Long M.Visualization of allostery in P-Selectin lectin domain using MD simulations.PLoS One, 2010, 5:e15417
[46].Stansfeld PJ, Sansom MS.Molecular simulation approaches to membrane proteins.Structure, 2011, 19:1562~1572
[47].Zhu F, Tajkhorshid E, Schulten K.Theory and simulation of water permeation in aquaporin-1.Biophys J, 2004, 86:50~57
[48].Zhang MH, LüSQ, Li G, Mao Z, Yu X, Sun WN, Tang Z, Long M, Su WA.Identification of a residue in helix 2 of rice plasma membrane intrinsic proteins that influences water permeability.J Biol Chem, 2010, 285:41982~41992
[49].Rapedius M, Fowler PW, Shang L, Sansom MS, Tucker SJ, Baukrowitz T.H bonding at the helix-bundle crossing controls gating in Kir potassium channels.Neuron, 2007, 55:602~614
[50].Sotomayor M, Vasquez V, Perozo E, Schulten K.Ion conduction through MscS as determined by electrophysiology and simulation.Biophys J, 2007, 92:886~902
[51].Stansfeld PJ, Hopkinson R, Ashcroft FM, Sansom MS.PIP (2) -binding site in Kir channels:Definition by multiscale biomolecular simulations.Biochem, 2009, 48:10926~10933
[52].Loura LM, Ramalho JP.Recent developments in molecular dynamics simulations of fluorescent membrane probes.Molecules, 2011, 16:5437~5452
[53].Fernandez-Recio J, Verma C.Theory and simulation:Integrating models into experimental scenarios.Curr Opin Struct Biol, 2010, 20:139~141
[54].Ranaghan KE, Mulholland AJ.Investigations of enzyme-catalysed reactions with combined quantum mechanics/molecular mechanics (QM/MM) methods.Inter Rev Phys Chem, 2010, 29:65~133
[55].Senn HM, Thiel W.QM/MM methods for biomolecular systems.Angew Chem Int Edit, 2009, 48:1198~1229
[56].Markwick PRL, McCammon JA.Studying functional dynamics in bio-molecules using accelerated molecular dynamics.Phys Chem Chem Phys, 2011, 13:20053~20065
[57].Zuckerman DM.Equilibrium sampling in biomolecular simulations.Annu Rev Biophys, 2011, 40:41~62
[58].Grant BJ, Gorfe AA, McCammon JA.Large conformational changes in proteins:Signaling and other functions.Curr Opin Struct Biol, 2010, 20:142~147
[59].Larsson P, Hess B, Lindahl E.Algorithm improvements for molecular dynamics simulations.Comput Mol Sci, 1:93~108
[60].Noid WG, Chu JW, Ayton GS, Krishna V, Izvekov S, Voth GA, Das A, Andersen HC.The multiscale coarse-graining method.I.A rigorous bridge between atomistic and coarse-grained models.J Chem Phys, 2008, 128:244114
[61].Noid WG, Liu P, Wang Y, Chu JW, Ayton GS, Izvekov S, Andersen HC, Voth GA.The multiscale coarse-graining method.II.Numerical implementation for coarse-grained molecular models.J Chem Phys, 2008, 128:244115
[62].Noe F, Horenko I, Schutte C, Smith JC.Hierarchical analysis of conformational dynamics in biomolecules:Transition networks of metastable states.J Chem Phys, 2007, 126:155102
[63].Walton EB, Vanvliet KJ.Equilibration of experimentally determined protein structures for molecular dynamics simulation.Phys Rev E, 2006, 74:061901
[64].Noe F, Doose S, Daidone I, Lollmann M, Sauer M, Chodera JD, Smith JC.Dynamical fingerprints for probing individual relaxation processes in biomolecular dynamics with simulations and kinetic experiments.Proc Natl Acad Sci USA, 2011, 108:4822~4827
[65].陈长军, 黄延昭, 肖奕.生物大分子的自由能计算方法.生物物理学报, 2011, 27 (5) :395~402 Chen CJ, Huang YZ, Xiao Y.Free energy calculation methods of biomolecules.Acta Biophys Sin, 2011, 27 (5) :395~402
[66].Durrant JD, McCammon JA.Molecular dynamics simulations and drug discovery.BMC Biol, 9:71~79





