
1 引 言
氘是自然界中最简单的原子之一, 常温下氘分子通过范德瓦耳斯力的作用结合在一起. 由于氘分子属于同核双原子分子, 不存在固有电子偶极距的变化, 因此常温下氘没有红外活性. 低温条件下, 相邻分子间的作用将促使氘分子电偶极距发生变化,因此液氘将表现出红外活性, 并且此时氘分子基本处于基态. 红外光谱测试技术以分子(原子)的振 (转) 动特性为基础. 国外学者已对低温下氘的红外吸收情况作了相关研究, 而目前国内尚未开展这方面的研究. 毕鹏等在实验上测得了三相点附近液体氢的红外吸收光谱, 并用非谐振子模型理论计算并解释了液氢的基频峰位置, 此方法仅计算了氢分子的振动情况而并未考虑分子的转动. 本文采用基于第一性原理的从头计算方法研究了D2d和D2h两种构型的基态(D2)3分子的能量模式以及红外谱图. 并且利用自主研制的低温平面冷冻靶系统和红外光谱测量系统获得了液氘的红外吸收谱.结果表明, 实验测得的液氘红外吸收谱的主吸收峰所在位置与理论计算得到的基态(D2)3分子的红外强度最强位置一致, 这说明处于基态的D2d和D2h两种构型的(D2)3分子能够模拟研究低温下液氘的红外吸收特性.
2 计算方法
本文计算是在基于第一性原理的单双取代耦合簇(CCSD)方法结合cc-PVTZ基组的框架下进行, 即所有计算均利用了CCSD理论并考虑了氘的3s, 2p, 1d价极化函数的基组. 计算过程首先对D2d结构以及D2h结构的(D2)3分子进行完全弛豫优化, 再进行频率分析并消去可能存在的虚频, 最后得到稳定的基态结构, 如图1 所示. 上述的所有计算均使用Gaussian03程序完成.
3 结果与讨论
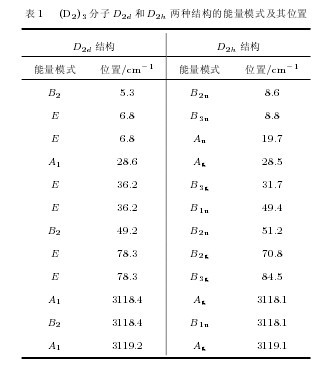
在完全弛豫优化的基础上, 计算得到了(D2)3分子最终的D2d稳定结构和D2h稳定结构. 在D2d结构中, 第一个氘分子和第二个氘分子位于同一平面, 第三个氘分子垂直于该平面, 三个氘分子的连线互成90°角. 在D2h结构中, 三个氘分子均在同一个平面, 分子相互连线互成90°角. 处于最低能量状态的这两种构型中氘分子的键长均为0.074 nm.表1给出了计算得到的基态(D2)3分子D2d和D2h两种结构的所有能量模式的对称性以及相应位置. 从表1可以发现, D2d构型的许多能量模式发生了简并. 基态(D2)3分子的能量模式主要分为低波数区和高波数区, 低波数区主要来源于(D2)3中分子的转动和平动, 高波数区主要以分子的振动为主. 在D2d结构中, 高波数区的能量模式有A1, B2, A1, 位置分别为3118.4, 3118.4,3119.2 cm. 而在D2h构型中, 这三个高波数区位置分别为 3118.1, 3118.1, 3119.1 cm1, 其对应的对称性分别是Ag, B1u, Ag.【表1】
据计算得到的基态(D2)3分子D2d和D2h两种结构的能量模式红外强度的差别, 我们分别做出了基态(D2)3分子的D2d构型和D2h构型的红外谱图, 结果如图2和图3所示.
由图2可知, 对于D2d结构的基态(D2)3分子,在低波数区出现的分立的能量模式红外强度相对较小, 而在高波数区出现的能量模式红外强度最大.比较图2和图3可以看出, D2h结构的基态(D2)3分子在低波数区的能量模式的红外强度要大于D2d结构在低波数区所表现出的能量模式的红外强度.
同样, D2h结构在高波数区的能量模式红外强度最大. 利用Av(J) 表示分子的振动能量跃迁和转动能量跃迁, 其中, v为跃迁后的振动量子态, J 为跃迁前的转动量子态, A为由J 的大小所决定的跃迁标记. 由于D2分子为同核双原子分子, 它的振 (转)动能量跃迁必然遵循能量跃迁基本定则: v = 0,±1; J = 0, ±2. 我们分别用Qv(J)和Sv(J)表示J = 0 和 J = ±2 的能量跃迁, Q1(0), Q1(1),S0(0), S0(1), S1(0), S1(1)分别表示v = 0 → 1,J = 0 → 0; v = 0 → 1, J = 1 → 1; v = 0 → 0,J = 0 → 2; v = 0 → 0, J = 1 → 3; v = 0 → 1,J = 0 → 2; v = 0 → 1, J = 1 → 3 的能量跃迁.
另外, 根据能量跃迁基本定则: J 等于2的整数倍时能量跃迁也会发生, 这里用Uv(J) 和Wv(J)表示J = 4和J = 6的能量跃迁, U0(0), U0(1),W0(0), W0(1) 分别表示v = 0 → 0, J = 0 → 4;v = 0 → 0, J = 1 → 5; v = 0 → 0, J = 0 → 6;v = 0 → 0, J = 1 → 7 的能量跃迁. 由于低温条件下D2分子几乎全部被限制在基态上, 因此D2分子发生振转能量跃迁时, 基本跃迁模式是由基态向第一激发态的跃迁. 对D2分子的振动能量跃迁而言, 最大概率跃迁方式为v = 0 → 1. 同时, 在低温下D2分子J = 1 的跃迁形式被限制, 因此D2分子转动能量跃迁的最大概率跃迁方式为J = 0 → 2. 由上述分析可知, 对于低温下液氘的能量跃迁, 振动方式以Q1(0)为主, 转动方式主要为S0(0), 振动和转动的共同作用Q1(0) + S0(0) 也是最有可能出现的跃迁模式.由表1计算结果并结合上述分子振转能量跃迁理论可知, 基态(D2)3分子的D2d结构和D2h结构表现出的红外强度最大位置3119.2, 3119.1 cm1均为Q1(0) + S0(0) 模式.
4 实验验证
液氘红外吸收测量实验全部在低温平面冷冻靶系统和红外光谱测量系统上完成. 关于该实验系统的具体的装置介绍见文献[17]. 首先利用低温平面冷冻靶系统, 在真空条件下 (真空度达到104Pa)把平面靶盒温度降至接近氘的三相点温度 (氘的三相点为18.71 K), 然后再以2.0 ml/min的进气速率向靶盒内充入氘气, 直至最后靶盒内充满液氘且整个系统状态稳定. 图4给出了通过红外成像获得的向平面靶盒中充入液氘以及充满液氘后系统最后处于一个稳定状态的过程.待充满液氘的靶盒处于一个稳定状态后 (系统处于恒定的温度场), 通过自主研制的低温红外光谱测量系统, 我们获得了低温条件下液氘的红外吸收谱, 结果如图5所示.从 图5可 以 发 现, 液 氘 在2900—3500 nm(3448—2857 cm1)波长范围内出现了一个强的吸收谱带, 其中在3160 cm处出现强吸收峰. 文献[21, 22]报道了伴有分子转动的固体氘的基频峰位置在3159 cm处, 由此可知本实验中液氘红外吸收谱中位于3160 cm处的主吸收峰对应的跃迁模式为Q1(0) + S0(0). 这与上述理论计算得到的D2d构型和D2h构型的基态(D2)3分子红外强度最大位置 (3119.2, 3119.1 cm) 基本一致. 液氘的能量模式Q1(0) + S0(0) 位置的实验结果和理论模拟计算结果列于表2.【表2】

由实验结果以及理论计算分析可知, 低温下液氘的能量跃迁以Q1(0)为主, 同时伴有分子的转动能量跃迁S0(0). 振动能量跃迁和转动能量跃迁的共同作用Q1(0) + S0(0) 模式不仅从理论上验证了低温下液氘的基本跃迁方式, 也验证了低温下氘分子大部分处于基态上, 同时说明低温下液氘的能量跃迁主要以低模式为主.
5 结 论
基于第一性原理的从头计算方法研究了处于基态的(D2)3分子的D2d构型和 D2h构型的能量模式, 根据不同模式红外强度的差异分别做出了基态(D2)3分子这两种构型的红外谱图. 通过自主研制的低温平面冷冻靶和红外光谱测量系统获得了液氘的红外吸收谱, 结果发现主吸收峰位置与理论计算得到的基态(D2)3分子这两种不同构型的红外强度最大位置一致. 这说明液氘的跃迁模式为Q1(0) + S0(0), 即低温下液氘的能量跃迁主要以低模式为主.





