
京都基因与基因组百科全书(Kyoto Encyclope-dia of Genes and Genomes, KEGG)中的通路数据库(KEGG PATHWAY database)是系统性分析和阐释基因功能的重要知识库, 涵盖了从基本细胞过程到人类复杂疾病等重要生命过程中分子间的相互作用和网络关系, 已成为研究细胞生化过程如代谢、膜转运、信号传递和细胞周期以及人类复杂疾病致病分子机制的重要参考工具.KEGG 通路通过描述分子间的相互互作和反应的信息以阐释基因及其产物的功能。KEGG 通路数据库中存储的数据对象也被称为是广义的蛋白质-蛋白质互作网络, 包括基因产物(节点)和 3 种类型的相互作用和关系(边):酶-酶关系、直接的蛋白质-蛋白质互作、基因表达关系.随着分子生物学研究的不断深入, KEGG 通路数据库得到快速地积累和扩充, 已从 2001 年的201 个通路约 6000 个基因产物节点增加到 2010 年的 300 多个通路约 17 000 多个蛋白质节点.尽管如此, 对于复杂的生命过程而言, 该数据库掌握的知识仍只是冰山一角, 亟需进一步进行知识扩充和完善。但是, 受限于实验成果发表周期以及数据库管理人员手工扩充通路信息造成的滞后, 传统的基于实验室技术的通路扩充方法已然不能满足当前分子生物学研究的需求。生物信息学预测方法为通路扩充提供了一种有效和便利的途径。
既往的通路(网络)扩充、重构和预测方法多基于基因表达数据提取基因间相互作用的关系.例如:Markus 等提出可以利用基于相关的方法扩充现有的调控网络; Luo 等利用基于三方互信息的方法推断转录调控网络。此外, 概率布尔网络和贝叶斯网络的方法也被广泛用于生物学通路的重构和扩充.然而, 这些方法存在以下几个缺陷:首先, 需要借助高通量基因表达谱数据, 这些数据往往不能满足理论模型依赖的统计分布假设条件; 其次, 这些方法没有充分利用到日益积累的蛋白质-蛋白质互作和基因功能注释等先验生物学知识, 其结果缺乏合理的生物学解释, 难以被广泛接受。
为了克服上述缺陷, 本文提出了一种基于蛋白质-蛋白质互作(Protein-protein interaction, PPI)和基因本体论(Gene Ontology, GO)数据库知识的通路扩充方法。该方法主要通过利用目标基因的互作邻居的功能学信息, 预测其可能参与的生物学通路以实现通路扩充的目的。PPI 数据库主要存储通过实验方法或者计算生物学方法获得的蛋白质-蛋白质互作信息, 已被广泛应用于分子网络的构建、功能分类以及基因功能预测等生物学研究。GO数据库是目前应用最广泛的基因功能注释体系之一,旨在建立基因及其产物知识的标准词汇体系, 从基因的细胞组分(Cellular component, CC)、分子功能(Molecular function, MF)和生物学过程 (Biologicalprocess, BP)3 个方面阐释基因的功能归属。
1 材料和方法
1.1 蛋白质互作数据来源
本文所用到的蛋白质-蛋白质互作数据来源于Human Protein Reference Database (HPRD)数据库和 Biological General Repository for Interaction Da-tasets (BioGRID)数据库.HPRD 数据库包含了利用体内、体外实验和酵母双杂交等技术获得的人类蛋白质-蛋白质互作知识, 涉及 9616 个人类基因间的 39 240 个互作对子。BioGRID 数据库包含了利用酵母双杂交实验获得的人类蛋白质-蛋白质互作关系, 涉及 12 582 个人类基因间的 101 925 个互作对子。为便于方法学评价, 本文对目标基因进行了以下筛选, 去掉符合下列条件之一的基因:(1)不能注释到任何 KEGG 通路; (2)不能注释到任何 GO 节点;(3)与其互作的基因不能注释到任何 KEGG 通路。经筛选, 在HPRD和BioGRID数据库中, 分别有3417个和3912 个人类基因纳入分析。本文利用 Bioconductor R软件包对基因进行 KEGG 和 GO 功能注释.
1.2 目标基因候选通路的识别
首先通过蛋白质-蛋白质互作信息确定与某一目标基因存在直接互作的邻居; 然后, 将互作邻居基因映射到 KEGG 数据库中, 查找其注释到的所有通路。目标基因的候选通路定义为与其存在蛋白质-蛋白质互作的所有邻居基因所能被注释到的一系列KEGG 通路。
1.3 目标基因的通路预测
蛋白质-蛋白质互作通常与特定的生物学途径有联系.研究表明, 相互作用的一对蛋白质倾向于共同参与特定的生物学过程, 因此 KEGG 通路可以看作一个广义的蛋白质-蛋白质互作网络。通路中的基因之间更倾向于存在较强的生物学关系, 它们往往共同参与特定的生物学过程并具有类似的生物学功能。因此, 如果一条通路中的基因倾向于富集在目标基因所注释到的 GO 节点上, 则可逆向推测该目标基因可能归属于此通路。给定一个目标基因和他的一个候选 KEGG 通路, 首先, 得到目标基因所注释到的 GO 节点列表; 其次, 针对每一个 GO 节点, 对候选通路中的所有基因(不包括目标基因)进行 GO 富集分析, 以 α = 0.05 为检验水准。对于一个GO 节点 A 和一个 KEGG 通路 B, 富集分析的 P 值可通过以下超几何分布公式计算:【1】
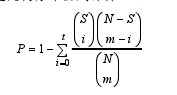
其中 t 是通路 B 中可以注释到 GO 节点 A 的基因个数, N 为 KEGG 数据库中的全部基因个数, m 为KEGG 数据库中所有能注释到 GO 节点 A 的基因个数, S 为通路 B 中的基因个数。最后, 考虑到 GO 涵盖了 3 个方面, 如果在目标基因所有的 BP 节点或者CC 节点或者 MF 节点显着富集, 则预测该目标基因属于此通路(称为预测通路)。
1.4 预测效果评估
参考文献[25], 本文采用两个指标评估预测效果, 分别是平均准确率(CR)和相对准确率(RP)。本文定义, 如果目标基因的预测通路中至少有一个与其已知的注释通路一致, 则称该基因可被成功预测。对于目标基因 k, Pk为其预测通路的集合, Tk为其已知的注释通路集合。假设有 n 个目标基因, 则 CR 值的计算公式如下:【2】
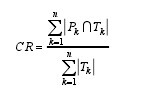
|·| 表示集合中的元素个数。CR 值越大, 表示方法的预测性能越好。
给定一个成功的预测, RP 值衡量该预测是完全正确(目标基因注释的所有通路都被预测正确)的可能性, 即预测的相对准确率。假设目标基因中有 l 个基因被成功预测, 其中s个被完全预测正确, 则RP = s/l.
对于 GO 功能节点的 BP、CC 和 MF 类, 本文设计了 4 种方案:GPPI-BP、GPPI-CC、GPPI-MF 和GPPI-BOCOM, 以探讨各 GO 功能类对预测结果的影响。GPPI-BP 只考虑 BP 节点, GPPI-CC 只考虑CC 节点; GPPI-MF 只考虑 MF 节点; GPPI-BOCOM综合了所有3种GO功能类的结果, 即只要在某一类节点达到富集即可。
2 结果与分析
2.1 4 种方案的预测结果
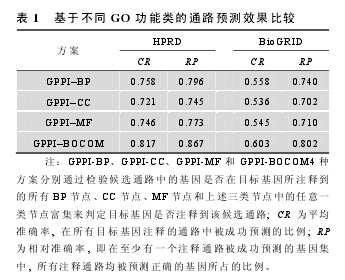
4 种方案的预测结果见表 1.基于 3 种 GO 功能类的方案(GPPI-BP、GPPI-CC 和 GPPI-MF)预测效果相近。在 HPRD 数据中, 3 种方案的 CR 值分别为75.8%、72.1%和 74.6%, RP 值分别为 79.6%、74.5%和 77.3%; 在 BioGRID 数据中, 3 种方案的 CR 值分别为 55.8%、53.6%和 54.5%, RP 值分别为 74.0%、70.2%和 71.0%.对比利用 BioGRID 数据进行预测的结果, 利用 HPRD 数据进行预测得到了更高的平均准确率和相对准确率。对 3 种方案进行比较发现,GPPI-BOCOM 的 预 测 效 果 最 好 , 在 HPRD 和BioGRID 数据中, CR 值分别为 81.7%和 60.3%, RP 值分别为 86.7%和 80.2%.因此, 后续仅报告基于GPPI-BOCOM 的结果。【表1】
2.2 互作邻居个数(k)对预测效果的影响
本文探讨了不同 k 值下对目标基因进行通路预测的效果。图 1给出了在不同k值下(k = 1, 2, …, 22)可被预测目标基因数目的分布以及部分预测正确和完全预测正确的基因数目的分布情况。在 HPRD 和BioGRID 两套数据中, 可被预测的目标基因的绝对数目均随互作邻居数目的增加而逐渐下降(分别从434 和 655(k = 1)减少到 32 和 35 (k = 22)), 而完全预测正确的基因个数占可被预测的目标基因的比例却呈现上升趋势, 说明互作邻居数目与目标基因的通路预测效果存在很强的关联。与 BioGRID 数据库相比, HPRD 数据库中可被预测的目标基因的绝对数目少一些, 但其中完全预测正确的基因比例却高许多, 尽管两个数据间的这些差异随着互作邻居个数的增加而逐渐消失。【图1】
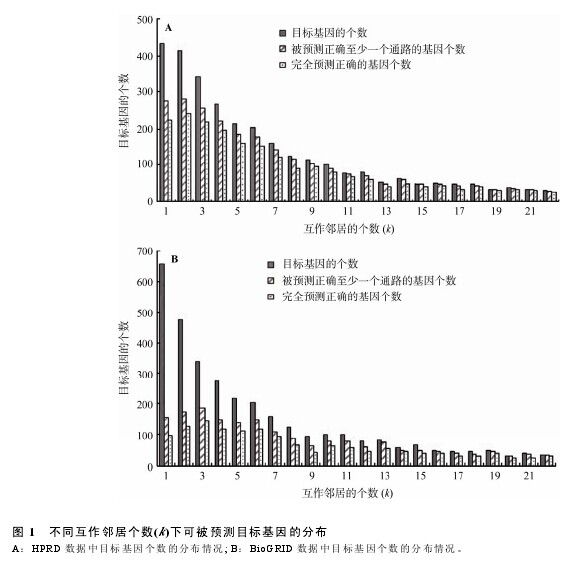
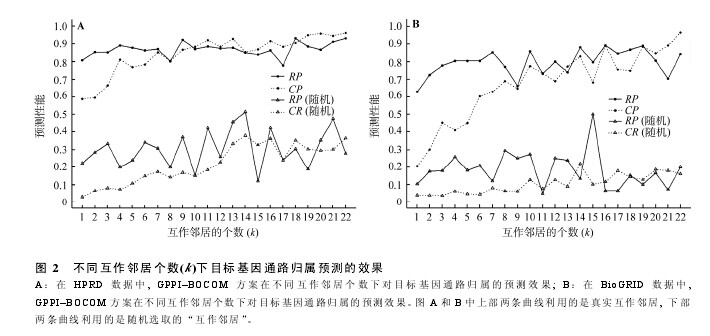
图 2 描绘了在不同 k 值下目标基因通路归属预测的变化趋势, 并与随机情况进行了对比。在两套数据中(图 2A 和 2B), CR 值及 RP 值均呈现持续上升的趋势, 但 BioGRID 的上升趋势更为明显。当互作邻居个数达到 22时, CR值分别达到了 96.2% (HPRD)和 96.3% (BioGRID), 而 RP 分别为 93.3%(HPRD)和84.1%(BioGRID)。值得注意的是, 在 HPRD 数据中当互作邻居个数 ≥ 5 时, CR 值已达到 90%.为了进一步评估提出的基于蛋白质-蛋白质互作知识的通路扩充方法的有效性, 本文与随机的预测方法进行了比较。对每个目标基因, 随机选取与实际互作邻居数目相同的基因(为避免混淆, 称为“互作邻居”),计算随机情况下的预测效果(其 RP 值和 CR 值随“互作邻居”个数的变化趋势见图 2A 和 2B 中下部的两条曲线)。在 HPRD 数据中, CR 值介于 2.9%~37.9%之间, RP值介于 12.0%~51.4%之间; 在 BioGRID数据中, CR 值介于 3.5%~21.9%之间, RP 值介于4.8%~50.0%之间。从图中可以看出, 随机情况下,两套数据中的 RP 值随“互作邻居”个数增大呈现小幅度的上升趋势, 而 CR 值无明显变化趋势。结果证明, 利用真实互作基因的预测效果要远好于利用随机挑选的基因的预测效果。【图2】
2.3 利用知识更新验证提出的通路预测方法的有效性
作为对预测方法进行验证的另外一种方式, 本文对新旧两个版本的 KEGG 通路数据库进行了数据收集, 分别为 2011 年 3 月 15 日发布的版本和 2012年 3 月 18 日发布的版本。定义在新版数据库中更新的基因通路为验证的对象。先用旧版数据库数据对被更新通路注释的基因进行预测, 然后根据新版数据库中的数据对其预测的正确性进行评估。新版数据库共更新了 89 个基因, 利用 HPRD 数据中的蛋白质-蛋白质互作知识, 提出的 GPPI 方法成功地预测了其中的 50 个基因(表 2), 其中 43 个基因的所有更新通路被全部预测出来, 预测的相对准确率为86.0%.从预测结果可以看出, 更新基因的部分预测通路在新版通路数据库中得到了验证, 且达到了较高的相对准确率, 证明了方法的有效性。【表2】
3 讨 论
本文提出了一种通过整合蛋白质-蛋白质互作知识和 GO 数据库对目标基因进行通路预测的新思路。基于 KEGG 通路是一个广义的蛋白质-蛋白质互作网络这一背景, 从蛋白质-蛋白质互作数据出发, 利用功能富集分析进行基因的通路预测。对利用蛋白质-蛋白质互作数据得到的目标基因的候选通路进行 GO 功能富集分析, 成功预测出了目标基因所注释到的部分或者全部通路, 达到了良好的预测效果。进一步利用新旧版本数据库的更新信息,对 KEGG 数据库中的更新基因进行预测, 部分预测结果在更新数据库中得到了验证, 从而证明了本文提出的方法的有效性和可靠性。通过与随机情况相比较, 我们较全面地评估了本文提出的方法的统计显着性。较之现有的基于基因表达谱数据及模型方法, 本方法的优势主要体现在以下几个方面:
第一, 不需要很强的理论假设。对于表达谱数据来说, 现有方法所作的假设有时并不一定能够得到满足, 造成通路预测的可靠性差; 第二, 本研究充分利用了蛋白质-蛋白质互作知识, 与 KEGG 通路的构建背景相吻合, 方法学更具有合理的生物学解释;第三, 本方法不需要事先定义一个基因族(或者子网)进行通路扩充, 避免了基因族(或者子网)定义的随意性; 第四, 从预测效果上看, 本文提出的方法显着优于之前基于数据采矿的方法。例如, Luo 等利用合成数据评估其提出的三方互信息法在推断转录调控网络关系的效果时, 正确率为 77.0%, 而基于 HPRD 数据, 当互作基因个数 ≥ 5 时, 本文提出的方法正确率达 90.0%, 其他方法多是根据基因归属于某个通路的可能性大小对基因进行排序。
从预测结果来看, BioGRID 数据的预测结果稍逊于 HPRD 数据的结果, 这可能是由如下原因造成的:BioGRID 数据库中的蛋白质-蛋白质互作仅通过酵母双杂交实验得到, 而 HPRD数据库的蛋白质-蛋白质互作是通过体内、体外和酵母双杂交实验中的至少一种实验得到的。因此, 与 HPRD 中的互作相比, BioGRID 的假阳性率更高。除本文所研究的KEGG 通路数据库外, 还有一些其他常用的通路数据库值得探索, 如 Reactome 和 BioCarta.Reactome通路数据库的基本单元是一个生化反应, 反应之间根据因果关系链组合起来形成生物途径来描述代谢、信号传导、DNA 修复和细胞周期调控等生物学过程, 已与 KEGG 数据库建立了广泛的交叉应用.
如何融合 Reactome 通路的构建背景和蛋白质-蛋白质互作知识对其进一步扩充将是我们进一步的研究方向。BioCarta 数据库在其公共网站上提供了用于绘制生物学通路的模板, 研究者可以把符合标准的生物学通路提供给 BioCarta 数据库, 但它不会检验这些生物学通路的质量, 故其中的资料质量参差不齐, 受数据库本身质量的影响, 对其进行预测的可靠性可能会降低。
作为一种探索性研究, 本研究受到数据库信息量完整性的影响。从预测结果可以看出, 大部分预测通路在更新的数据库中得到了证实, 但仍有部分预测通路未被现有的知识所证实。然而, 这些新发掘的通路很可能是进行生物学通路归属预测的价值所在, 为探索生物学通路未知的空间提供了一个行之有效的方法, 同时也为进一步开展湿实验验证研究指明了方向。当然, 在实际预测时可通过整合几种分子互作数据库得到可信度更高更完善的蛋白质-蛋白质互作证据, 以达到更好更可靠地预测效果。
本方法具有很好的推广性, 可以应用于其他类型的分子互作数据分析以及对其他生物的 KEGG 通路扩充研究中。此外, 本方法另一个不足之处是将每个GO 节点同等对待。事实上, 一些 GO 节点之间是存在紧密联系的, 形成了一种层次结构, 在未来的研究中我们也将进一步利用层次结构信息研发预测效能更佳的算法。
参考文献
[1] Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M.KEGG for integration and interpretation of large-scalemolecular data sets. Nucleic Acids Res, 2012, 40(Databaseissue): D109–D114.
[2] Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genesand genomes. Nucleic Acids Res, 2000, 28(1): 27–30.
[3] Chen L, Zhang LC, Zhao Y, Xu LD, Shang YK, Wang Q,Li W, Wang H, Li X. Prioritizing risk pathways: a novelassociation approach to searching for disease pathwaysfusing SNPs and pathways. Bioinformatics, 2009, 25(2):237–242.
[4] Lee E, Chuang HY, Kim JW, Ideker T, Lee D. Inferringpathway activity toward precise disease classification.PLoS Comput Biol, 2008, 4(11): e1000217.
[5] Li Y, Agarwal P. A pathway-based view of human diseasesand disease relationships. PLoS ONE, 2009, 4(2): e4346.
[6] Kanehisa M, Goto S, Kawashima S, Nakaya A. The KEGGdatabases at GenomeNet. Nucleic Acids Res, 2002, 30(1):42–46.
[7] Kanehisa M. The KEGG database. Novartis Found Symp,2002, 247: 91–101, discussion 101–103, 119–128, 244–152.





