摘 要: 酿酒酵母Saccharomyces cerevisiae是代谢工程中最重要的宿主之一,先进的基因编辑技术已经被广泛应用于酿酒酵母细胞工厂的设计和构建。随着基因编辑技术的飞速发展,早期基于重组酶和同源重组的基因编辑技术逐渐被新型基因编辑系统所替代。文中对酿酒酵母基因编辑技术的原理和应用进行了总结,包括经典的酿酒酵母基因编辑技术,基于核酸内切酶的MegNs、ZFNs和TALENs等基因组编辑系统,最后介绍和讨论了基于CRISPR/Cas系统、异源代谢途径多拷贝整合和基因组规模基因编辑的最新研究进展,并对酿酒酵母基因编辑技术的应用前景和发展方向进行了展望。
关键词: 酿酒酵母; 基因编辑; CRISPR; 多拷贝整合;
Abstract: Saccharomyces cerevisiae is one of the most important hosts in metabolic engineering. Advanced gene editing technology has been widely used in the design and construction of S. cerevisiae cell factories. With the rapid development of gene editing technology, early gene editing technologies based on recombinase and homologous recombination have been gradually replaced by new editing systems. In this review, the principle and application of gene editing technology in S. cerevisiae are summarized. Here, we first briefly describe the classical gene editing techniques of S. cerevisiae. Then elaborate the genome editing system of MegNs, ZFNs and TALENs based on endonuclease. The latest research progress is especially introduced and discussed, including the CRISPR/Cas system, multi-copy integration of heterologous metabolic pathways, and genome-scale gene editing. Finally, we envisage the application prospects and development directions of Saccharomyces cerevisiae gene editing technology.
Keyword: Saccharomyces cerevisiae; gene editing; CRISPR; multi-copy integration;
为实现生物能源、精细化学品和天然产物等的绿色、可持续制造,利用代谢工程与合成生物学方法构建细胞工厂是一种有前途的策略[1]。作为一种典型的真核生物系统,酿酒酵母Saccharomyces cerevisiae具有优良的鲁棒性、对苛刻发酵条件的耐受性、基因工程操作的高效性和公认的安全性(Generally recognized as safe,GRAS),在学术研究和工业发酵中已经得到了广泛应用[2,3,4]。早期酿酒酵母主要用于发酵面制品(如面包、馒头、包子)和酒精饮料(如啤酒、米酒)等的生产,这一特性被很快用于燃料乙醇和单细胞蛋白的发酵生产。随着代谢工程技术的发展,酿酒酵母被越来越多的用于生产有机酸、氨基酸、核苷酸、药用蛋白、工业酶制剂和多不饱和脂肪酸(PUFAs)[5]等,这些产品被广泛应用于食品、医药、饲料和化工行业[6]。此外,酿酒酵母在天然产物的微生物异源合成方面也显示出了独特的优势。与原核生物相比,酿酒酵母具有较为完整的翻译后修饰系统和细胞器系统(例如线粒体、过氧化物酶体、内质网、高尔基体和液泡),为天然产物的生物合成提供了不同的环境和间隔[7,8],特别是在异源表达复杂代谢途径以及表达膜结合蛋白细胞色素P450氧化酶时表现出优越的能力[9,10,11,12,13]。目前,多种植物次生代谢产物已经在酿酒酵母中实现合成,如生物碱[11]、芳香氨基酸[14]、萜类[15]和黄酮类[16]等。最成功的例子是抗疟药物青蒿素前体物青蒿酸在酿酒酵母中的合成,产量高达25 g/L,并已实现工业化生产[17]。

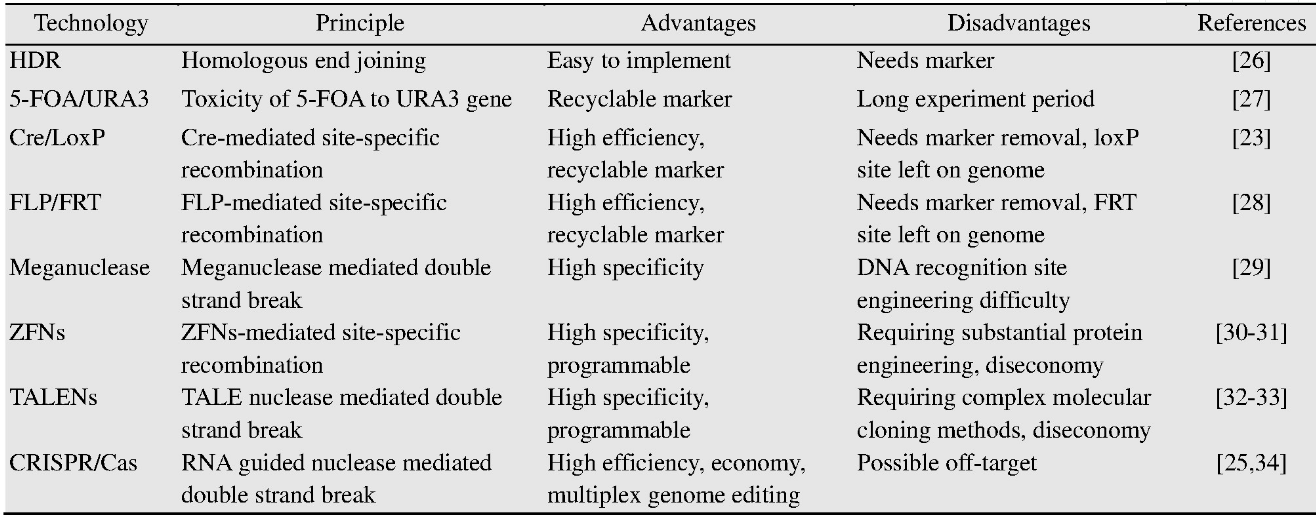
“设计-构建-测试-学习”是代谢工程的一般过程和实现细胞工厂构建预期目标的必经之路[18](图1)。其中,构建是验证设计的必要手段。然而,随着越来越多的化合物在酿酒酵母中实现合成,细胞工厂的构建与途径优化过程也暴露出了缺陷和局限,例如需要在单一位点进行多次操作[14],这已成为代谢工程的限速步骤。因此,开发高效的基因编辑工具势在必行。基因编辑技术是对目标基因组位点进行特异性修饰,包括基因敲除、敲入、替换和有义的点突变[19]。基因编辑技术在现代生物技术中起到至关重要的作用,其要求对基因和基因组预定位置进行精确高效的编辑和修饰[20,21]。传统的基因编辑技术是根据DNA同源重组(Homologous recombination,HR)原理,利用外源供体基因片段敲除或敲入目标基因片段,但这种方法效率较低,发生基因重组的概率在10–9–10–6[22]。酿酒酵母中常用的基因编辑系统是Cre/Lox P系统,同样是基于同源重组原理的基因编辑技术[23]。为了提高基因重组的效率,基因组双链断裂已经被证明有助于提高同源重组发生的概率[24,25]。基于此,新型基因编辑技术已经被开发利用,主要包括4种:归巢核酸内切酶(Meganucleases,Meg Ns)系统、锌指核酸酶(Zinc finger nucleases,ZFNs)系统、转录激活因子样效应物核酸酶(Transcription activator-like effector nucleases,TALENs)系统和CRISPR (Clustered regularly interspaced short palindromic repeats)相关的系统(表1)[19]。这些新技术的开发和应用,极大地促进了代谢工程与合成生物学的发展。本文对酿酒酵母基因编辑技术的发展和现状进行了总结,包括早期基因编辑技术、新型编辑技术和高通量编辑技术等。并对酿酒酵母基因编辑技术的发展前景进行了展望。
图1 酿酒酵母细胞工厂构建
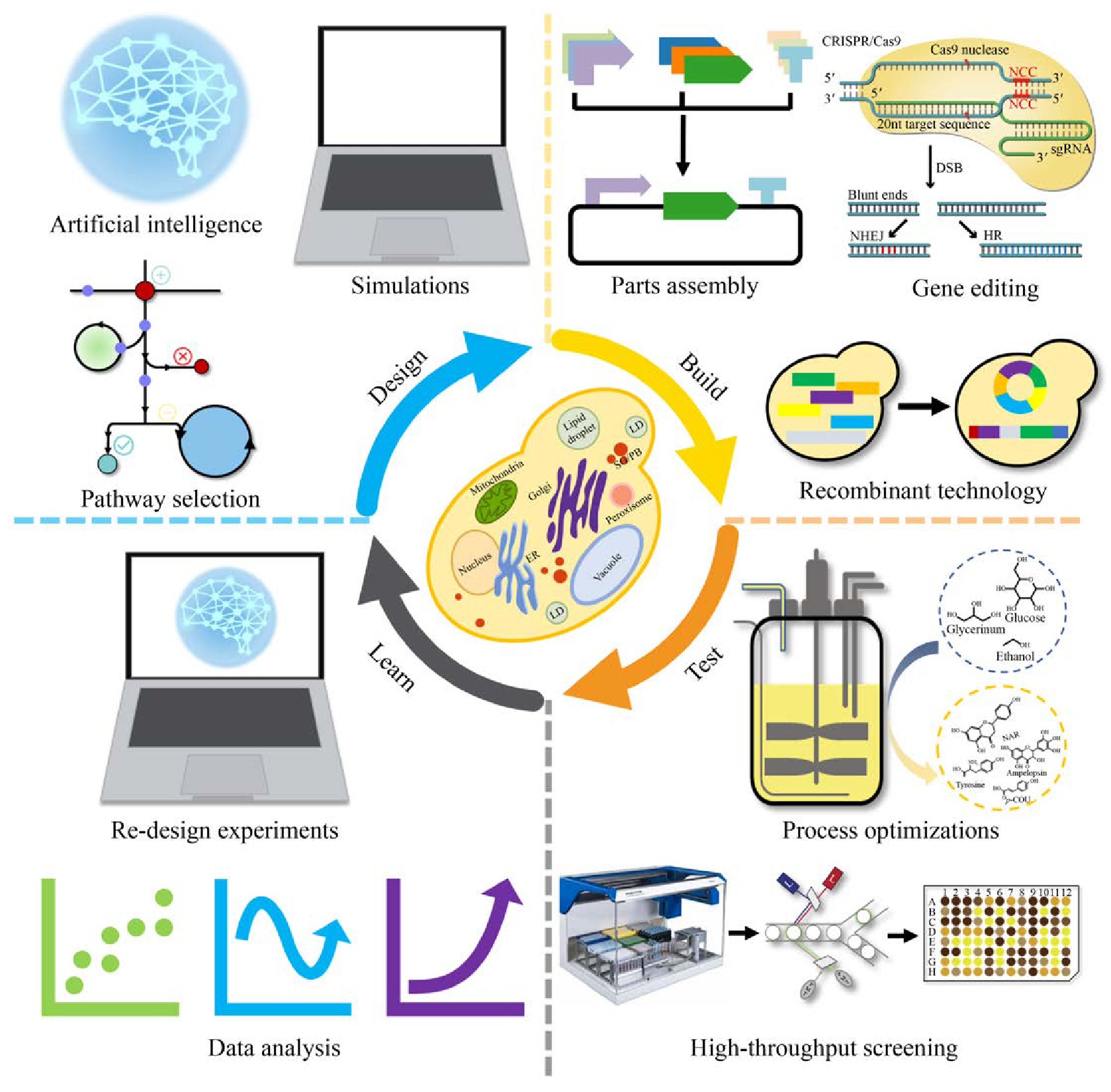
Fig.1 Construction of S.cerevisiae cell factory.
表1 基因编辑技术在酿酒酵母中的应用
1 、酿酒酵母经典基因编辑技术
20世纪70年代,基因工程的发展为基因组编辑开辟了新途径[35]。早期酿酒酵母基因编辑技术主要基于同源重组和筛选标记。其中,同源重组技术利用未损伤的同源DNA片段作为模板,以实现目标基因的添加、替换或失活[36]。这种经典的基因组编辑技术已在细胞工厂构建中得到了广泛应用。在酿酒酵母中主要存在两种机制参与DNA双链断裂修复:同源重组(Homology-directed repair,HDR)(图2A)和非同源末端连接(Nonhomologous end-joining,NHEJ)(图2B)[24]。HDR是利用带有目标基因同源序列的DNA对目标位点实现精确的基因编辑。而NHEJ则是细胞自身在一些修复元件的作用下随机的修复过程,这可能造成基因组碱基的插入和缺失,导致基因开放阅读框突变[37]。
随后,可以实现选择标签循环利用的技术被开发,包括5-FOA/URA3负筛选技术和基于重组酶的负筛选系统。其中,5-FOA/URA3负筛选技术利用5-氟乳清酸(5-fluoroorotic acid,5-FOA)对URA3阳性酿酒酵母工程菌株具有毒性的特点,来筛选URA3阴性酿酒酵母工程菌株(图2C)[38]。基于重组酶作用的选择标签回收系统同样已经被广泛应用到微生物细胞工程的构建中[39],包括来源于酿酒酵母2μ质粒的特异性重组系统FLP/FRT系统[28]和源自噬菌体P1的Cre/lox P系统[40](图2D)。与5-FOA/URA3负筛选技术相比,这两种系统具有更高的编辑效率。但这两种方法都依赖反向重复序列(Inverted repeats,IRs),发生重组之后会在基因组中留下一个重复序列的副本(lox P或FRT位点),在多次重复回收选择标签后导致基因组的不稳定性增加[41]。为了避免冗余重复序列对后续基因编辑的影响,经突变的FRT和lox P位点可避免这种情况[42]。尽管上述系统很大程度上消除了筛选标签可用性的限制,但由于缺乏在单一转化步骤中同时引入多个基因修饰的可靠方法,使用筛选标签替代靶基因仍然是一个耗时的过程[43]。此外,还需在工程菌株中表达相应的重组酶,或者需要携带相应重组酶基因的质粒进行额外转化并在随后的实验中消除。虽然这些方法的出现提高了细胞工厂构建的效率,但需使用筛选标签对基因组进行连续修改,并且对基因组进行多目标编辑时并不理想。
图2 经典基因编辑技术
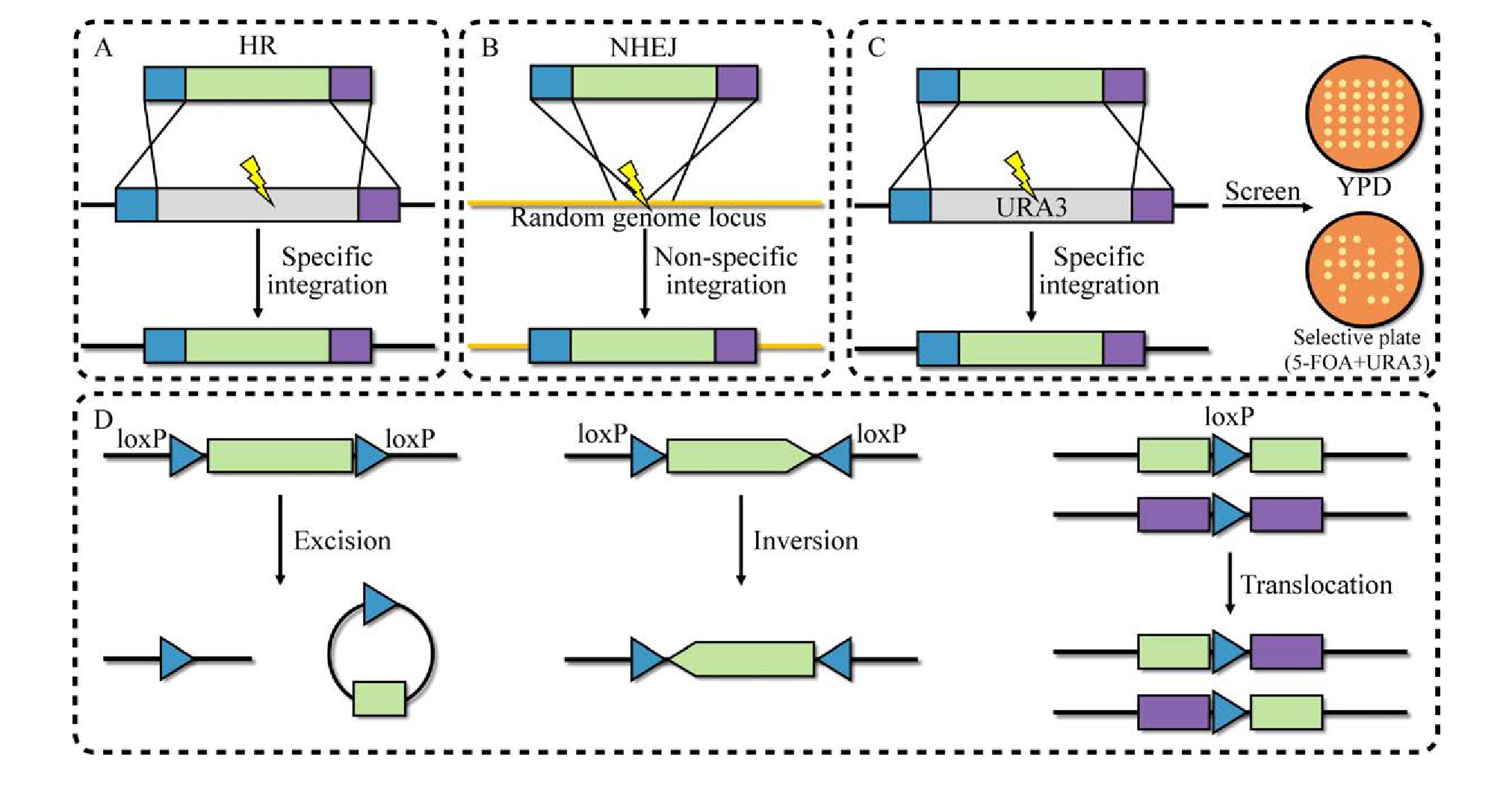
Fig.2 Classic gene editing technology.(A) Homologous recombination connection.(B) Non-homologous terminal connection.(C) 5-FOA/URA3 negative screening technology.(D) Cre/lox P technology.
2、 基于Meg Ns、ZFNs和TALENs的基因组编辑系统
随着基因编辑技术的迅猛发展,早期基因编辑系统已经逐渐被新型基因编辑系统替代。基于核酸内切酶的基因编辑技术包括归巢核酸内切酶(Meg Ns)技术、锌指蛋白核酸酶(ZFNs)技术[44]、转录激活因子样效应物核酸酶(TALENs)[45]技术和CRISPR相关技术[46]等。通过对目标基因序列造成双链断裂(Double strand break,DSB),进而对其进行定点基因组编辑。由于DSB在酵母中具有致命性,因此这些方法理论上可用于无筛选标签修饰,并对目标位点进行精确改造。
2.1、 Meg Ns技术
Meg Ns是具有较大识别位点特征的脱氧核糖核酸内切酶,其可识别超过14个碱基对的双链DNA序列,如酿酒酵母中的Ⅰ-SceⅠ归巢核酸内切酶可以识别18 bp的序列[47]。为了实现对目标基因的编辑,通常需要将归巢核酸内切酶识别位点先引入宿主的染色体目标位置,随后Meg Ns可在基因组目标位点附近引入DSB,进而提高基因编辑效率。Kuijpers等利用Ⅰ-SceⅠ在目标染色体位点引入双链断裂,使基因片段的整合效率提高到95%[41]。
2.2、 ZFNs技术
ZFNs技术是指由锌指核酸酶介导的基因编辑技术,锌指核酸酶是较早用于基因组编辑的人工合成限制性内切酶,主要由两部分组成包括:限制性核酸内切酶FokⅠ的非特异性DNA切割结构域和可识别特异性DNA的锌指蛋白(Zinc finger protein,ZFP)(图3)[48]。ZFP由串联的Cys2-His2锌指蛋白模块构成,通常单个Cys2-His2锌指由大约30个氨基酸组成,为一个α-螺旋和2个反向的β平行构成β-β-α结构[48]。适当增加锌指结构关联数可以更准确地识别目标序列,进而提高锌指蛋白对基因组目标片段的特异性识别[49]。每个锌指蛋白模块可以选择性识别DNA序列的3个碱基对(Base pair,bp),并通过其α-螺旋残基与DNA大沟(Major groove of DNA)相互作用,形成碱基特异性接触[50]。FokⅠ需要形成二聚体才可以靶向基因组中的目标序列以进行有效的基因编辑。因此,通常需要两分子ZFNs才能与DNA以适合的方向结合并定位到目标序列,这使特异性识别的碱基数量增加了一倍[50]。
图3 基于核酸内切酶的基因编辑技术
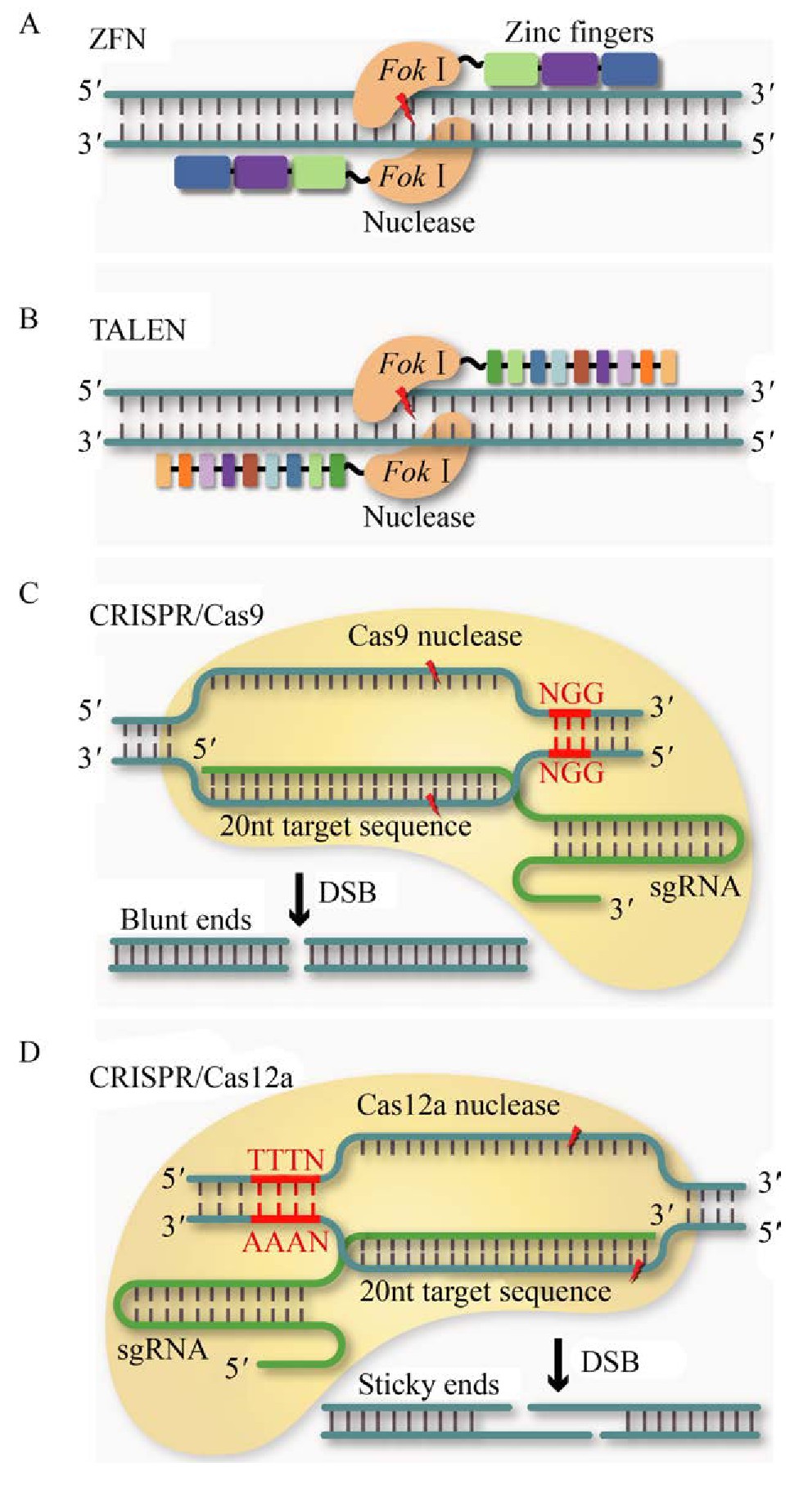
Fig.3 Gene editing technology based on endonuclease.(A) ZFN system (B) TALEN system (C) CRISPR/Cas9system (D) CRISPR/Cas12a system.
酿酒酵母作为经典的模式微生物,已经被用作验证新设计ZFNs的效率[47]。将丧失功能的MEL1报告基因整合到酵母基因组中,随后利用携带候选ZFNs的质粒测试编辑效率[30]。具有活性的ZFNs对目标DNA造成双链断裂,最后通过同源重组的方式修复报告基因[30]。
2.3、 TALENs技术
TALENs同样是一种工程化核酸酶,包含一个非特异性的DNA切割域和一个DNA特异性结合域(图3)。与ZFNs技术类似,也需要利用限制性核酸内切酶FokⅠ的非特异性DNA切割结构域对基因组进行编辑[51]。DNA特异性结合域由转录激活因子样效应因子(TALE)高度保守的重复序列组成,最初在植物黄单胞菌Xanthomonas campestris中被发现[52]。TALE的重复结构域通常由34个氨基酸残基组成,其中第12个和第13个重复序列可变双氨基酸残基(Repeat-variable di-residues,RVDs)决定了DNA结合的特异性[53]。每个RVD可以识别一个特定的核苷酸,从而形成一个固定的DNA识别代码(NI为腺嘌呤;HD为胞嘧啶;NG为胸腺嘧啶;NH或NN为鸟嘌呤),其可以按顺序组装以结合任何基因组目标序列[54]。不同的RVD与FokⅠ相连接形成TALENs,可识别的目标序列长度通常为14–20 bp,2个FokⅠ可以形成二聚体进而对基因组进行编辑。与ZFNs相比,TALENs的构建过程相对简单,并且应用范围更广,灵活性和效率也更高[55]。但由于其成本相对高昂,至今尚无一种低成本的且能公开的获取方法来快速产生大量的TALENs。在酿酒酵母中,利用ZFNs和TALENs技术对基因组目标位点造成双链断裂后,可通过内源HR或NHEJ修复机制实现对目标基因位点的定点编辑。然而,对不同基因进行修饰时,必须设计和重新合成ZFNs或TALENs蛋白。
3 、基于CRISPR的基因组编辑技术
CRISPR/Cas基因编辑技术是2013年出现的由小分子RNA介导的一种靶向基因组编辑的新技术。已成为代替ZFNs和TALENs技术诱导靶向基因编辑的一种潜在简便而有效的方案[56]。目前,CRISPR/Cas系统已经成为世界上最流行的基因组编辑工具,已应用在各种生物体中,包括细菌、真菌、植物和哺乳动物[24]。CRISPR/Cas系统是一种适应性免疫系统,源于细菌和古菌中用来抵御外来核酸的免疫防御机制[57]。CRISPR基因座已经在大约40%的细菌和90%的古菌物种中发现[58]。CRISPR/Cas系统由多个Cas基因的操纵子和一组非编码CRISPR-RNAs (cr RNAs)组成。cr RNA包括被短重复序列间隔分开的特异性重复序列。特异性重复序列是从外来DNA中获得的特殊基因片段,针对外来DNA序列的特异性识别。当发生DNA入侵时,新的特异识别序列也可以被整合到cr RNA中,保护自身不会再次被同一外来DNA干扰[59,60]。
3.1、 CRISPR基因组编辑技术原理及分类
CRISPR/Cas系统对外来DNA的切割主要分3个阶段,包括外源特异DNA俘获、cr RNA合成和切割瓦解[57,61]。首先,CRISPR/Cas系统的一个Cas蛋白子集相关基因对入侵的外来DNA序列进行识别,并将入侵DNA序列作为特异性重复序列插入到宿主基因组CRISPR阵列部分,以及复制一个新的CRISPR阵列。它的识别基于其序列下游的前间区序列邻近基序(Protospacer adjacent motif,PAM),不同来源的CRISPR系统的PAM序列也存在差异。随后,CRISPR阵列被转录并加工成单个的cr RNA,每个cr RNA都带有与先前入侵的外来DNA序列相对应的RNA片段以及部分CRISPR重复序列。最后,cr RNA引导由Cas蛋白组成的复合物或单个蛋白对入侵DNA序列进行切割瓦解。
CRISPR/Cas系统具有多样性,包括以DNA为靶点的系统、以RNA为靶点的系统和以DNA与RNA为靶点的系统[57]。根据其效应蛋白的数量可以将CRISPR/Cas系统分为两大类,1类(包括Ⅰ、Ⅲ和Ⅳ型)是使用多蛋白复合物;2类(Ⅱ型、Ⅴ型和Ⅵ型)利用单个核酸酶(如Cas9、Cas12和Cas13)。1类和2类CRISPR/Cas系统在生化结构上分别具有相似的特点。Ⅰ型、Ⅲ型和Ⅳ型是由4–7个Cas蛋白亚基组成多蛋白效应复合物。Ⅱ型、Ⅴ型和Ⅵ型则由单独多结构域蛋白组成。近年来,随着对该系统结构和功能的不断深入探索研究,CRISPR系统已成为一种成熟高效的基因编辑工具,其中应用最为广泛的是CRISPR/Cas9系统和新兴的CRISPR/Cas12a(Cpf1)系统。
3.2、 CRISPR/Cas9系统
以来源于酿脓链球菌Streptococcus pyogenes的Ⅱ型CRISPR/Cas9系统为例,其是对抗外来入侵DNA的防御系统[39]。该系统是由Cas9蛋白、cr RNA和tracr RNA (trans-activating cr RNA)构成的复合体[47](图3)。Cas9蛋白包含两个核酸酶结构域HNH和Ruv C,它们分别切割与cr RNA中20个核苷酸(nt)引导序列互补和非互补的DNA链,形成平末端DNA双链断裂[62]。经人工改造后,cr RNA和tracr RNA被组合成为一个g RNA(guide RNA)分子。通过碱基配对作用,自折叠成部分双链的RNA结构,与Cas9结合发挥功能。因此,仅需要通过改变g RNA与目标基因组DNA特异性结合序列便可以实现基因编辑的目的。靶向目标序列的另一个要求是同样需要PAM区域以使Cas9蛋白识别结合。Cas9蛋白复合体在临近PAM位点区域对目标序列进行剪切产生DSB。随后通过酿酒酵母中的同源修复机制,便可以实现基因组目标基因的缺失或整合。
3.3、 CRISPR/Cas9系统在酿酒酵母中的应用
CRISPR/Cas9系统已经成为酿酒酵母基因组编辑的革命性和多功能策略。目前,CRISPR系统已被开发为具有基因敲除、整合、转录干扰和转录激活的能力,代谢工程中的大多数基因操作都可以利用CRISPR/Cas9系统实现[63]。CRISPR/Cas9系统通过工程化的单链引导RNA (Single guide RNA,sg RNA)引导Cas9核酸酶效应体形成复合物,从而切割PAM序列上游3 bp处的基因组,造成DSB[64]。当供体DNA存在与基因组DNA同源序列时,酿酒酵母便可通过DSB实现对基因组的特异性基因编辑。
Di Carlo等首次报道了利用CRISPR/Cas系统对酿酒酵母基因进行敲除[65]。在没有筛选标签的情况下,通过90 bp同源序列实现了CAN1基因的敲除。Jako?iūnas等开发了用于酿酒酵母基因组多基因敲除的CRISPR/Cas9系统,最多可以同时对5个不同的基因组位点进行敲除[66]。通过共转化g RNA质粒和相应的线性同源重组片段,单个基因至5个基因的编辑效率达到50%–100%,工程菌株甲羟戊酸产量比野生型菌株提高了41倍。Eau Claire等成功将17个编码β-胡萝卜素合成的基因片段一次性整合到酿酒酵母基因组中,相邻DNA片段仅有50 bp互补,再一次证明了CRISPR/Cas9系统与同源重组相结合的有效性[67]。Zhang等将t RNA序列与多个g RNA串联构建了g RNA-t RNA-array CRISPR/Cas9 (GTR-CRISPR)系统,同时敲除8个基因的效率达到87%,极大地提高了基因编辑效率[20]。
对于工业酿酒酵母而言,基因操作可能更加复杂。同时,对于用于食品发酵工业或者酿造工业的酿酒酵母,从食品安全的角度出发,此类酿酒酵母不应携带有抗性标签和毒性成分编码基因,因此对其遗传修饰需要更加严谨,并通过严格的安全性评价后方可获批上市。然而,多个等位基因的存在和缺乏筛选标记加大了遗传操作的难度[68]。因此,CRISPR/Cas9系统的无标记编辑方式更适合应用到二倍体和多倍体工业菌株的基因改造中[69]。Lee等利用CRISPR/Cas9系统构建了工业酵母菌株JHS200的营养缺陷型突变体,并将其应用于利用银草Silver grass水解液生产生物乙醇的研究中[68]。通过引入木糖途径和NADH氧化酶提高了纤维素乙醇的生产,最终乙醇产量达到55.5 g/L[68]。Zhang等利用CRISPR/Cas9系统在工业多倍体酿酒酵母ATCC 4124中实现了营养标签URA3、TRP1、LEU2和HIS3的敲除[70]。Lian等通过增加表达g RNA质粒的拷贝数提高了基因敲除效率,敲除二倍体菌株和三倍体菌株中4个基因的效率为100%[71]。这些研究表明,CRISPR/Cas9系统在多倍体工业株的应用是可行的。
CRISPR/Cas9系统不仅仅是具有基因编辑的功能,经工程化的CRISPR/Cas9系统还可以实现其他多重功能,包括基因的抑制和激活[63]。对Cas9核酸酶结构域HNH (H840A)和Ruv C(D10A)进行突变,可导致两个结构域失活从而使核酸酶丧失切割活性[72,73,74]。如果仅突变其中一个位点,可以使Cas9蛋白成为切口酶。其只切割双链DNA中的一条链,当与一对g RNA联合使用时,可分别在两条目标DNA链上产生切口。
3.4、 CRISPR/Cas12a(Cpf1)系统
CRISPR/Cas12a(Cpf1)是一种新型基因编辑技术,属于依赖于单组分效应蛋白干扰基因的2类Ⅴ型CRISPR系统[75](图3)。以来源于氨基酸球菌Acidaminococcus spp.BV3L6的As Cpf1和来源于毛螺科菌Lachnospiraceae bacterium ND2006的Lb Cpf1为例,Cas12a识别的PAM位点为目标DNA 5’端处富含T的序列(如5’-TTTN-3’),在g RNA引导下与目标DNA特定位点结合并执行切割功能[75]。与Sp Cas9相比,As Cpf1和Lb Cpf1的分子量更小,更容易进入细胞,且只需要cr RNA无需tracr RNA即可引导切割,具有切割DNA和RNA的能力[76]。此外,剪切方式与Cas9也有较大差异。Cas9是在同一个位置同时剪切DNA分子的双链,最后形成平末端,而Cas12a(Cpf1)剪切后形成两个不同长度的链并形成黏性末端[75]。同时,更长的PAM序列提高了识别目标序列的能力,使CRISPR/Cas12a(Cpf1)系统脱靶率更低[77]。Li等使用CRISPR/Cas12a(Cpf1)系统成功删除了酿酒酵母基因组上两个基因之间长达38 kb的片段,证明CRISPR/Cas12a(Cpf1)系统可用于酿酒酵母的基因组简化[78]。Verwaal等研究了3种不同来源的Cas12a(Cpf1)对酿酒酵母基因组编辑的功能,其中的氨基酸球菌属Acidaminococcus spp.BV3L6和新凶手弗朗西丝菌Francisella novicida U112两个来源的Cas12a(Cpf1)在单基因编辑上具有与CRISPR/Cas9系统相当的编辑效率,此外,来源于毛螺科菌Lachnospiraceae bacterium ND2006的Lb Cas12a(Cpf1)可实现高效的多重基因组编辑,可同时将3个类胡萝卜素基因表达框整合到3个不同的基因组位点[79]。
由上可以看出,不论对于CRISPR/Cas9系统还是CRISPR/Cas12a(Cpf1)系统来说,sg RNA的引导作用都是至关重要的。CRISPR系统已广泛应用在动物、植物和微生物的基因编辑中。然而,CRISPR系统的精确基因编辑一直受脱靶效应困扰。sg RNA的稳定结合能力和特异性在CRISPR系统中起到了重要作用。因此,获得高质量的sg RNA是CRISPR编辑成功的关键条件之一。
3.5 、优化sg RNA的设计
为了降低脱靶效应,基于计算机分析的sg RNA分析工具已经被开发以提高CRISPR系统的精确度[80]。但是,由于不同酿酒酵母基因组碱基存在一定差异,sg RNA设计软件或网站基因组数据存在局限性,在一定程度上限制了CRISPR系统的编辑效率。因此,获得针对性的基因组碱基数据同样十分重要。此外,将Cas9切口酶与双sg RNA相结合策略,同样可以降低脱靶效率[81]。需要注意的是,两条sg RNA需要足够近,且要定位在两条链上,以对目标基因造成双链断裂。此外,基于sg RNA文库和多功能CRISPR的系统(Multi-functional genome-wide CRISPR,MAGIC)已经被用来对目标基因进行精确编辑和调控基因表达水平[46]。
3.6 、碱基编辑器
碱基编辑器(Base editor,BE)是利用丧失活性的Cas9 (d Cas9或Cas9切口酶)与胞苷脱氨酶(Cytidine deaminases)或腺苷脱氨酶融合,对基因组进行单碱基突变的技术[82]。利用sg RNA的靶向能力,可以在不引入DNA双链断裂的情况下对目标碱基进行有针对性的编辑。丧失活性的Cas9与胞苷脱氨酶融合,可将脱氧胞苷脱氨为脱氧尿苷,使基因组上原有的C·G转变为T·A。与脱氧腺苷脱氨酶(Deoxyadenosine deaminase)融合,可将脱氧尿苷脱氨形成脱氧胞苷,使基因组上原有的A·T转变为G·C。利用碱基编辑器的靶向DNA诱变已经在酿酒酵母中得到证实[83]。但是,碱基编辑器也存在一定的局限性,其中最为显着的问题是目前只能用于不同碱基之间的转换(Transition),而无法实现不同碱基之间的颠换(Transversion),即它只能实现嘧啶对嘧啶和嘌呤对嘌呤的改变,而无法完成嘧啶对嘌呤和嘌呤对嘧啶的改变。另外一点就是碱基编辑器在RNA水平以及全基因组水平上存在较高的脱靶效应。
4 、异源代谢途径多拷贝整合
基于单拷贝位点的异源基因整合方式已经应用到酿酒酵母基因组整合中[84]。然而,由于拷贝数的限制,异源基因的表达水平和目标产物产量也可能被限制。利用酿酒酵母自身多拷贝位点的整合方式可进一步提高外源基因的拷贝数,更适合应用于代谢工程的改造中[85]。酿酒酵母中常用作异源基因整合的多拷贝位点主要有核糖体DNA (Ribosome DNA,r DNA)、δ位点和Ty转座子(图4)。
r DNA单元由两个转录区(5S和35S)和两个非转录间隔(NTS1和NTS2)组成,一个r DNA单元长度大约为9.1 kb,大约由150个拷贝组成[86]。基于r DNA位点的整合方式,已被广泛应用到工程菌株的构建中。Dai等[87]首先将来源于西洋参Panax quinquefolius的Pg DDS、Pg PPDS和拟南芥Arabidopsis thaliana的At CPR1基因整合到菌株BY4742的r DNA位点实现了原人参二醇的合成,并在后续实验中稳定表达。Zhang等[88]将β-香树脂醇合成途径以及强化底物供应途径整合到r DNA位点,工程菌株SGib实现了β-香树脂醇的高水平合成。随后,Zhu等[15]继续在工程菌株SGib上进行改造,将两个细胞色素P450酶(CYP72A154、CYP88D6)和一个细胞色素P450还原酶ATR1的基因整合到基因组上,成功合成了甘草次酸。Park等[89]组装胞质异丁醇生物合成途径到r DNA位点,q PCR分析表明3拷贝途径基因被整合,异丁醇产量达227.2 mg/L,多次传代后异丁醇产量基本一致,表明异源途径基因可以稳定整合在r DNA位点。
图4 酿酒酵母中异源代谢途径多拷贝整合
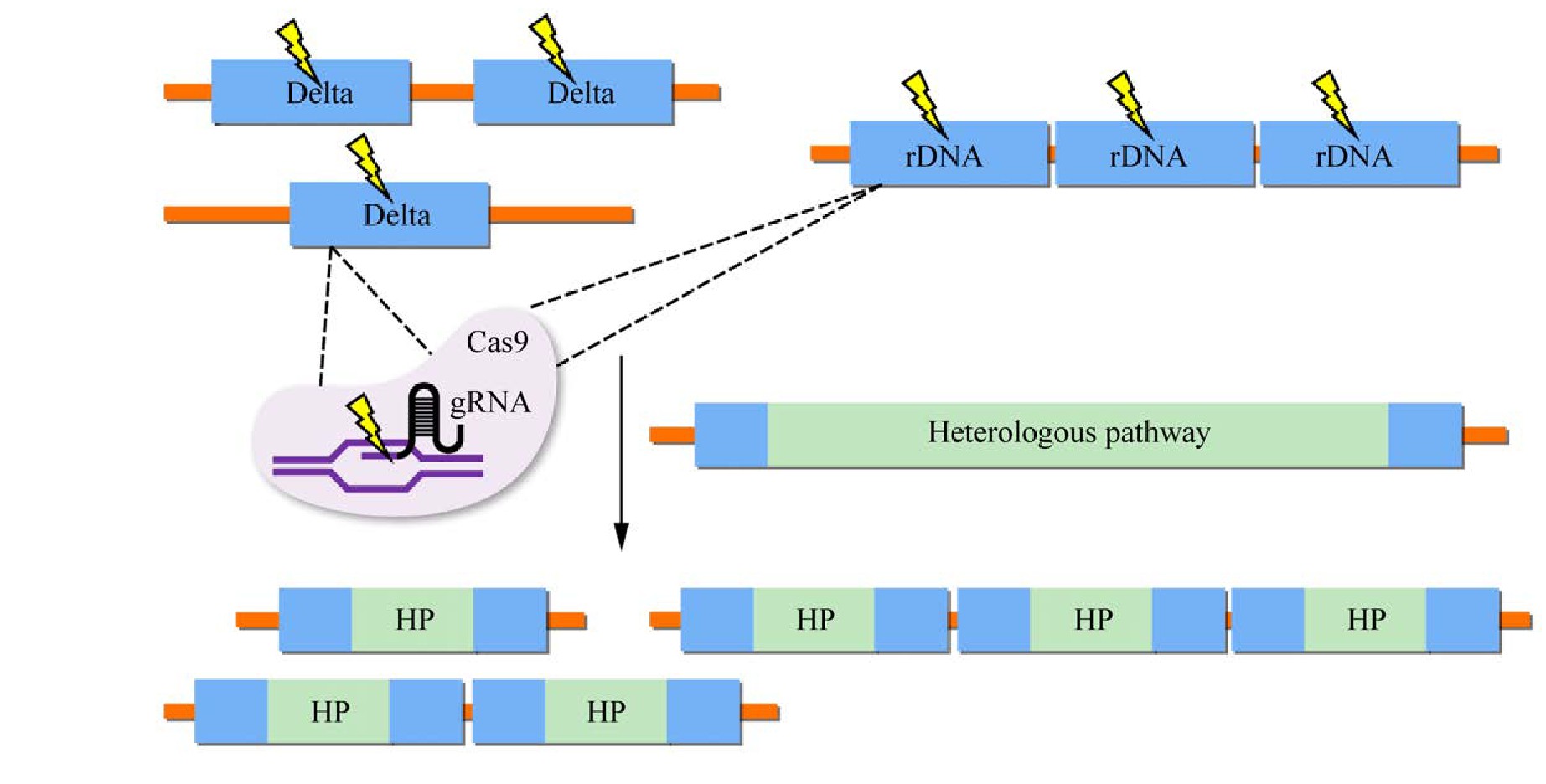
Fig.4 Multi-copy integration of heterologous metabolic pathways in S.cerevisiae.
酿酒酵母中存在具有转座能力的Ty因子(Ty elements),每段序列长度均在6 kb左右,两端分别接有长约0.33 kb的同向重复序列。Ty因子被分为五大家族,即Ty1到Ty5,且在酵母基因组上拷贝数较多,可作为外源基因的插入位点[60]。Shi等利用Di-CRISPR系统将木糖消耗途径和(R,R)-2,3-丁二醇生物合成途径,整合到Ty转座子的δ序列中,实现了大型异源生物合成途径的高效无标记整合。该系统通过对基因组δ位点产生DSB,最多整合了18个拷贝的24 kb DNA片段,研究表明较高的途径拷贝数提高了木糖的消耗率和(R,R)-2,3-丁二醇的产量[25]。以上研究表明,将异源途基因整合到酿酒酵母基因组高拷贝位点具有巨大潜力,这为异源途径整合奠定了基础。
5 、基因组规模的基因编辑
由于酿酒酵母代谢网络十分复杂,单基因或多基因的编辑可能限制了目标产物的生产。借助高通量基因组编辑技术可以实现多基因的自动基因组改造。此外,酿酒酵母基因组和单条染色体酿酒酵母的合成,为更深刻理解基础生命科学提供了参考。
5.1 、高通量基因编辑技术
从自然样本中筛选分离获得的微生物,由于目标化合物产量低和对恶劣工业条件耐受性弱等因素,通常很少能直接用于工业规模生产。因此,需要对微生物进行基因组水平的改造和进化,以满足工业化需求。然而,传统改造方法由于通量低限制了细胞工厂的构建。随着自动化设备和基因组编辑技术的发展,多种高通量筛选(Highthroughput screening,HTS)方法被开发,以高通量形式在基因组水平上修改细胞生理功能来重建细胞工厂,为菌株生长和产物积累提供最佳条件,最终提高细胞工厂的生产性能[90,91,92]。
对微生物基因组进行大规模高通量改造,可以获得巨大的突变库,再经过高通量筛选便可以实现特定目标化合物的生产。为了更合理地构建菌株文库,目前已经建立了基于随机组合多重自动基因工程(Multiplex automated genome engineering,MAGE)的高通量基因编辑技术[93]。最初,MAGE被应用在对大肠杆菌染色体上多个基因位点的快速连续改造,以实现大规模编程和进化[94]。随后,类似MAGE的酵母寡聚介导基因组工程(Yeast Oligo-Mediated Genome Engineering,YOGE)和真核生物多重基因组工程(Eukaryotic multiplex genome engineering,e MAGE)也被应用于酿酒酵母靶向工程化改造中[95,96]。e MAGE可在不导致DSB的情况下对酿酒酵母的多基因实现精确编辑修饰。通过在酿酒酵母中引入与复制叉的后随链互补的寡核苷酸,实现了大于40%的序列修饰效率[96]。多次循环同时靶向基因内的多个位点提高了工程菌株突变效率,该方法产生了近100万个突变体。对β-胡萝卜素途径的启动子、编码序列和终止子进行编辑,获得了β-胡萝卜素产量不同的工程菌株。此外,基于CRISPR系统的Cas EMBLR技术可将同源序列的目标DNA片段整合到基因组的特定位点,实现酿酒酵母体内多基因无标记组装[97]。Liu等设计了一种组合重编程基因表达以控制复杂表型的方法(Multiplex navigation of global regulatory networks,MINR),并构建了MINR文库[98]。通过乙醇和/或葡萄糖耐受性实验,特异性突变体不仅提高了乙醇和/或葡萄糖耐量,乙醇的生产浓度也比野生型菌株提高了2倍。Guo等基于CRISPR/Cas9系统创建了一个全基因组文库,在不同的环境条件下,对315个特征较差的小开放阅读框的重要性进行了评估[99]。
5.2 、人工合成基因组
DNA合成和组装方法的发展,促进了染色体的人工构建和全基因组重排,使病毒、生化途径、细菌和真菌基因组的人工合成成为可能[100]。“人工合成酿酒酵母基因组”计划(Sc2.0)是一个国际项目,旨在设计和构建一个完全合成的酵母基因组[101]。2011年,Dymond等证明了化学合成酿酒酵母Ⅸ号染色体右臂(synⅨR)和Ⅵ号染色体左臂(synⅥL)的可行性[102]。2014年,Annaluru等成功设计组装了第一个完整酿酒酵母染色体Ⅲ号染色体(synⅢ),使Ⅲ号染色体长度精简到272 871 bp[100]。到目前为止,Sc2.0联盟成员已经完成了synⅡ[103]、synⅤ[104]、synⅥ[105]、synⅩ[106]和synⅫ[107]染色体的构建。此外,单条染色体酿酒酵母的人工构建,对探索生命起源与进化同样具有重要价值[108]。酿酒酵母基因组设计和重构赋予了细胞工厂全新功能和进化潜力,为更全面探索一个有机体提供了可能[101,108]。
6、 总结与展望
酿酒酵母作为真核生物已经实现了化学品、植物次级代谢产物和异源蛋白质等的合成。尽管这些产物十分重要,但目前大部分基于酿酒酵母的产品的生产水平仍未达到商业化标准。为了实现目标产物的高水平生物合成,通常需要涉及几十个酶促反应。因此,引入的异源代谢途径与酿酒酵母之间的协调性尤为关键。需要考虑到异源途径的表达、代谢负担、前体物供应和次级代谢物生产强度对宿主的影响等因素。虽然酿酒酵母已经是研究最深入的真核微生物,但对整个代谢和调节网络仍然缺乏清晰而透彻的理解。通过基因编辑技术的进一步发展,各个基因功能及代谢网络的解析必将使人们对酵母世界的认知得到进一步的提升。
新型基因编辑工具的快速发展,已经极大地提高了酿酒酵母基因组工程的速度和效率。与传统基因编辑相比,基于CRISPR系统的技术在基因编辑和异源代谢途径组装等方面已显示出优势,可以在无筛选标记情况下同时进行多基因编辑,极大地减少了异源代谢途径引入和基因定点突变的周期,使单个基因或基因代谢网络组合优化成为可能。虽然,基于CRISPR系统的基因编辑技术还存在一些问题,如:当需要对目标基因进行改造时,往往需要设计多条特异性序列,以保证可以实现准确的基因编辑。因此,如何提高基因编辑的效率和精度成为最重要的任务。但是,随着CRISPR系统的进一步发展,更多Cas蛋白的开发利用使该技术在今后的农业、工业和医学领域中具有更广阔的应用前景。
此外,在工程构建过程中需要对底盘细胞进行多次改造,以使细胞工厂生产目标产物的效率和产量达到最佳水平,对如何利用人工智能等计算机辅助手段来提升改造的可靠性和效率提出了更高要求。基因组规模的基因编辑工程与机器学习的交叉融合或将在此领域大放异彩。由于多次改造必然会导致时间周期变长和经济消耗,如何在单次实验中同时完成大量的基因编辑工作,也是限制细胞工厂构建速度的关键因素。
参考文献
[1] Mougiakos I,Bosma EF,De Vos WM,et al.Next generation prokaryotic engineering:the CRISPR-cas toolkit.Trends Biotechnol,2016,34(7):575-587.
[2] Qin JF,Zhou YJ,Krivoruchko A,et al.Modular pathway rewiring of Saccharomyces cerevisiae enables high-level production of L-ornithine.Nat Commun,2015,6:8224.
[3] B?er E,Steinborn G,Kunze G,et al.Yeast expression platforms.Appl Microbiol Biotechnol,2007,77(3):513-523.
[4] Lyu XM,Zhao GL,Ng KR,et al.Metabolic engineering of Saccharomyces cerevisiae for de novo production of kaempferol.J Agric Food Chem,2019,67(19):5596-5606.
[5] Veen M,Lang C.Production of lipid compounds in the yeast Saccharomyces cerevisiae.Appl Microbiol Biotechnol,2004,63(6):635-646.
[6] Lian JZ,Chao R,Zhao HM.Metabolic engineering of a Saccharomyces cerevisiae strain capable of simultaneously utilizing glucose and galactose to produce enantiopure (2R,3R)-butanediol.Metab Eng,2014,23:92-99.
[7] Xu XH,Liu YF,Du GC,et al.Microbial chassis development for natural product biosynthesis.Trends Biotechnol,2020,38(7):779-796.
[8] Paddon CJ,Keasling JD.Semi-synthetic artemisinin:a model for the use of synthetic biology in pharmaceutical development.Nat Rev Microbiol,2014,12(5):355-367.
[9] Yang JZ,Liang JC,Shao L,et al.Green production of silybin and isosilybin by merging metabolic engineering approaches and enzymatic catalysis.Metab Eng,2020,59:44-52.
[10] Li MJ,Kildegaard KR,Chen Y,et al.De novo production of resveratrol from glucose or ethanol by engineered Saccharomyces cerevisiae.Metab Eng,2015,32:1-11.
[11] Galanie S,Thodey K,Trenchard IJ,et al.Complete biosynthesis of opioids in yeast.Science,2015,349(6252):1095-1100.
[12] Siddiqui MS,Thodey K,Trenchard I,et al.Advancing secondary metabolite biosynthesis in yeast with synthetic biology tools.FEMS Yeast Res,2012,12(2):144-170.
[13] Suastegui M,Guo WH,Feng XY,et al.Investigating strain dependency in the production of aromatic compounds in Saccharomyces cerevisiae.Biotechnol Bioeng,2016,113(12):2676-2685.
[14] Liu QL,Yu T,Li XW,et al.Rewiring carbon metabolism in yeast for high level production of aromatic chemicals.Nat Commun,2019,10:4976.
[15] Zhu M,Wang CX,Sun WT,et al.Boosting11-oxo-β-amyrin and glycyrrhetinic acid synthesis in Saccharomyces cerevisiae via pairing novel oxidation and reduction system from legume plants.Metab Eng,2018,45:43-50.
[16] Lv YK,Xu S,Lyu YB,et al.Engineering enzymatic cascades for the efficient biotransformation of eugenol and taxifolin to silybin and isosilybin.Green Chem,2019,21(7):1660-1667.
[17] Paddon CJ,Westfall PJ,Pitera DJ,et al.High-level semi-synthetic production of the potent antimalarial artemisinin.Nature,2013,496(7446):528-532.
[18] Palazzotto E,Tong YJ,Lee SY,et al.Synthetic biology and metabolic engineering of actinomycetes for natural product discovery.Biotechnol Adv,2019,37(6):107366.
[19] Yang ZL,Blenner M.Genome editing systems across yeast species.Curr Opin Biotechnol,2020,66:255-266.
[20] Zhang YP,Wang J,Wang ZB,et al.A g RNA-t RNAarray for CRISPR-Cas9 based rapid multiplexed genome editing in Saccharomyces cerevisiae.Nat Commun,2019,10:1053.
[21] Simon AJ,D’Oelsnitz S,Ellington AD.Synthetic evolution.Nat Biotechnol,2019,37(7):730-743.
[22] Hsu PD,Lander ES,Zhang F.Development and applications of CRISPR-Cas9 for genome engineering.Cell,2014,157(6):1262-1278.
[23] Lv XM,Wang F,Zhou PP,et al.Dual regulation of cytoplasmic and mitochondrial acetyl-Co Autilization for improved isoprene production in Saccharomyces cerevisiae.Nat Commun,2016,7:12851.
[24] Li L,Liu XC,Wei KK,et al.Synthetic biology approaches for chromosomal integration of genes and pathways in industrial microbial systems.Biotechnol Adv,2019,37(5):730-745.
[25] Shi SB,Liang YY,Zhang MM,et al.A highly efficient single-step,markerless strategy for multi-copy chromosomal integration of large biochemical pathways in Saccharomyces cerevisiae.Metab Eng,2016,33:19-27.
[26] Shao ZY,Zhao H,Zhao HM.DNA assembler,an in vivo genetic method for rapid construction of biochemical pathways.Nucleic Acids Res,2009,37(2):e16.
[27] Gao S,Lyu YB,Zeng WZ,et al.Efficient biosynthesis of (2S)-naringenin from p-coumaric acid in Saccharomyces cerevisiae.J Agric Food Chem,2020,68(4):1015-1021.
[28] Park YN,Masison D,Eisenberg E,et al.Application of the FLP/FRT system for conditional gene deletion in yeast Saccharomyces cerevisiae.Yeast,2011,28(9):673-681.
[29] Chevalier BS,Stoddard BL.Homing endonucleases:structural and functional insight into the catalysts of intron/intein mobility.Nucleic Acids Research,2001,29(18):3757-3774.
[30] Doyon Y,Mc Cammon JM,Miller JC,et al.Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases.Nat Biotechnol,2008,26(6):702-708
[31] Kim YG,Cha J,Chandrasegaran S.Hybrid restriction enzymes:zinc finger fusions to FokⅠcleavage domain.Proc Natl Acad Sci USA,1996,93(3):1156-1160.
[32] Zhang GQ,Lin YP,Qi XN,et al.TALENs-assisted multiplex editing for accelerated genome evolution to improve yeast phenotypes.ACS Synth Biol,2015,4(10):1101-1111.
[33] Christian M,Cermak T,Doyle EL,et al.Targeting DNA double-strand breaks with TAL effector nucleases.Genetics,2010,186(2):757-761.
[34] Vincent JJ,Martin,Leanne,et al.A highly characterized synthetic landing pad system for precise multi-copy gene integration in yeast.ACSsynth biol,2018,7(11):2675-2685.
[35] Rothstein RJ.One-step gene disruption in yeast.Methods Enzymol,1983,101:202-211.
[36] Schultz C,Lian JZ,Zhao HM.Metabolic engineering of Saccharomyces cerevisiae using a trifunctional CRISPR/Cas system for simultaneous gene activation,interference,and deletion.Methods Enzymol,2018,608:265-276.
[37] Bibikova M,Carroll D,Segal DJ,et al.Stimulation of homologous recombination through targeted cleavage by chimeric nucleases.Mol Cell Biol,2001,21(1):289-297.
[38] Ko N,Nishihama R,Pringle JR.Control of 5-FOAand 5-FU resistance by Saccharomyces cerevisiae YJL055W.Yeast,2008,25(2):155-160.
[39] 李诗渊,赵国屏,王金.合成生物学技术的研究进展--DNA合成、组装与基因组编辑.生物工程学报,2017,33(3):343-360.Li SY,Zhao GP,Wang J.Enabling technologies in synthetic biology-DNA synthesis,assembly and editing.Chin J Biotech,2017,33(3):343-360 (in Chinese).
[40] Gueldener U,Heinisch J,Koehler GJ,et al.Asecond set of lox P marker cassettes for Cre-mediated multiple gene knockouts in budding yeast.Nucleic Acids Res,2002,30(6):e23.
[41] Solis-Escalante D,Van Den Broek M,Kuijpers NGA,et al.The genome sequence of the popular hexose-transport-deficient Saccharomyces cerevisiae strain EBY.VW4000 reveals Lox P/Cre-induced translocations and gene loss.Fems Yeast Res,2015,15(2):fou004.
[42] Storici F,Lewis LK,Resnick MA.In vivo site-directed mutagenesis using oligonucleotides.Nat Biotechnol,2001,19(8):773-776.
[43] Mans R,Van Rossum HM,Wijsman M,et al.CRISPR/Cas9:a molecular Swiss army knife for simultaneous introduction of multiple genetic modifications in Saccharomyces cerevisiae.FEMSYeast Res,2015,15(2):fov004.
[44] Carroll D.Genome engineering with zinc-finger nucleases.Genetics,2011,188(4):773-782.
[45] Mussolino C,Morbitzer R,Lütge F,et al.A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity.Nucleic Acids Res,2011,39(21):9283-9293.
[46] Lian JZ,Schultz C,Cao MF,et al.Multi-functional genome-wide CRISPR system for high throughput genotype-phenotype mapping.Nat Commun,2019,10:5794.
[47] David F,Siewers V.Advances in yeast genome engineering.FEMS Yeast Res,2015,15(1):1-14.
[48] 刘耀,熊莹喆,蔡镇泽,等.基因编辑技术的发展与挑战.生物工程学报,2019,35(8):1401-1410.Liu Y,Xiong YZ,Cai ZZ,et al.Development and challenges of gene editing technology.Chin JBiotech,2019,35(8):1401-1410 (in Chinese).
[49] 张白雪,孙其信,李海峰.基因修饰技术研究进展.生物工程学报,2015,31(8):1162-1174.Zhang BX,Sun QX,Li HF.Advances in genetic modification technologies.Chin J Biotech,2015,31(8):1162-1174 (in Chinese).
[50] Li HY,Yang Y,Hong WQ,et al.Applications of genome editing technology in the targeted therapy of human diseases:mechanisms,advances and prospects.Signal Transduct Target Ther,2020,5:1.
[51] Li T,Huang S,Jiang WZ,et al.TAL nucleases(TALNs):hybrid proteins composed of TALeffectors and FokⅠDNA-cleavage domain.Nucleic Acids Res,2011,39(1):359-372.
[52] Boch J,Scholze H,Schornack S,et al.Breaking the code of DNA binding specificity of TAL-TypeⅢeffectors.Science,2009,326(5959):1509-1512.
[53] Montague TG,Cruz JM,Gagnon JA,et al.CHOPCHOP:a CRISPR/Cas9 and TALEN web tool for genome editing.Nucleic Acids Res,2014,42(W1):W401-W407.
[54] Streubel J,Blücher C,Landgraf A,et al.TALeffector RVD specificities and efficiencies.Nat Biotechnol,2012,30(7):593-595.
[55] Wei CX,Liu JY,Yu ZS,et al.TALEN or Cas9-rapid,efficient and specific choices for genome modifications.J Genet Genomics,2013,40(6):281-289.
[56] Gaj T,Gersbach CA,BarbasⅢCF.ZFN,TALEN,and CRISPR/Cas-based methods for genome engineering.Trends Biotechnol,2013,31(7):397-405.
[57] Zhang F.Development of CRISPR-Cas systems for genome editing and beyond.Quart Rev Biophys,2019,52:e6.
[58] Gupta D,Bhattacharjee O,Mandal D,et al.CRISPR-Cas9 system:A new-fangled dawn in gene editing.Life Sci,2019,232:116636.
[59] Barrangou R,Fremaux C,Deveau H,et al.CRISPRprovides acquired resistance against viruses in prokaryotes.Science,2007,315(5819):1709-1712.
[60] Jensen NB,Strucko T,Kildegaard KR,et al.Easy Clone:method for iterative chromosomal integration of multiple genes Saccharomyces cerevisiae.FEMS Yeast Res,2014,14(2):238-248.
[61] Shmakov S,Smargon A,Scott D,et al.Diversity and evolution of class 2 CRISPR-Cas systems.Nat Rev Microbiol,2017,15(3):169-182.
[62] Nishimasu H,Ran FA,Hsu PD,et al.Crystal structure of Cas9 in complex with guide RNA and target DNA.Cell,2014,156(5):935-949.
[63] Lian JZ,Hamedi Rad M,Hu SM,et al.Combinatorial metabolic engineering using an orthogonal tri-functional CRISPR system.Nat Commun,2017,8:1688.
[64] Gasiunas G,Barrangou R,Horvath P,et al.Cas9-cr RNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria.Proc Natl Acad Sci USA,2012,109(39):E2579-2586.
[65] Di Carlo JE,Norville JE,Mali P,et al.Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems.Nucleic Acids Res,2013,41(7):4336-4343.
[66] Jako?iūnas T,Bonde I,Herrg?rd M,et al.Multiplex metabolic pathway engineering using CRISPR/Cas9in Saccharomyces cerevisiae.Metab Eng,2015,28:213-222.
[67] Eau Claire SF,Zhang JZ,Rivera CG,et al.Combinatorial metabolic pathway assembly in the yeast genome with RNA-guided Cas9.J Ind Microbiol Biotechnol,2016,43(7):1001-1015.
[68] Lee YG,Jin YS,Cha YL,et al.Bioethanol production from cellulosic hydrolysates by engineered industrial Saccharomyces cerevisiae.Bioresour Technol,2017,228:355-361.
[69] Stovicek V,Borodina I,Forster J.CRISPR/Cas system enables fast and simple genome editing of industrial Saccharomyces cerevisiae strains.Metab Eng Commun,2015,2:13-22.
[70] Zhang GC,Kong II,Kim H,et al.Construction of a quadruple auxotrophic mutant of an industrial polyploid Saccharomyces cerevisiae strain by using RNA-guided Cas9 nuclease.Appl Environ Microbiol,2014,80(24):7694-7701.
[71] Lian JZ,Bao HZ,Hu SM,et al.Engineered CRISPR/Cas9 system for multiplex genome engineering of polyploid industrial yeast strains.Biotechnol Bioeng,2018,115(6):1630-1635.
[72] Gilbert LA,Horlbeck MA,Adamson B,et al.Genome-scale CRISPR-mediated control of gene repression and activation.Cell,2014,159(3):647-661.
[73] Jinek M,Chylinski K,Fonfara I,et al.Aprogrammable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity.Science,2012,337(6096):816-821.
[74] Ran FA,Hsu PD,Lin CY,et al.Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity.Cell,2013,154(6):1380-1389.
[75] Zetsche B,Gootenberg JS,Abudayyeh OO,et al.Cpf1 is a single RNA-guided endonuclease of a Class 2 CRISPR-Cas System.Cell,2015,163(3):759-771.
[76] Fonfara I,Richter H,Bratovi?M,et al.The CRISPR-associated DNA-cleaving enzyme Cpf1also processes precursor CRISPR RNA.Nature,2016,532(7600):517-521.
[77] 杨帆,李寅.新一代基因组编辑系统CRISPR/Cpf1.生物工程学报,2017,33(3):361-371.Yang F,Li Y.The new generation tool for CRISPRgenome editing:CRISPR/Cpf1.Chin J Biotech,2017,33(3):361-371 (in Chinese).
[78] Li ZH,Liu M,Lyu XM,et al.CRISPR/Cpf1facilitated large fragment deletion in Saccharomyces cerevisiae.J Basic Microbiol,2018,58(12):1100-1104.
[79] Verwaal R,Buiting-Wiessenhaan N,Dalhuijsen S,et al.CRISPR/Cpf1 enables fast and simple genome editing of Saccharomyces cerevisiae.Yeast,2018,35(2):201-211.
[80] 李红,谢卡斌.植物CRISPR基因组编辑技术的新进展.生物工程学报,2017,33(10):1700-1711.Li H,Xie KB.Recent progresses in CRISPRgenome editing in plants.Chin J Biotech,2017,33(10):1700-1711 (in Chinese).
[81] Cho SW,Kim S,Kim Y,et al.Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases.Genome Res,2014,24(1):132-141.
[82] Komor AC,Badran AH,Liu DR.Editing the genome without double-stranded DNA breaks.ACSChem Biol,2018,13(2):383-388.
[83] Nishida K,Arazoe T,Yachie N,et al.Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems.Science,2016,353(6305):aaf8729.
[84] Lyu XM,Ng KR,Lee JL,et al.Enhancement of naringenin biosynthesis from tyrosine by metabolic engineering of Saccharomyces cerevisiae.J Agric Food Chem,2017,65(31):6638-6646.
[85] Lian JZ,Mishra S,Zhao HM.Recent advances in metabolic engineering of Saccharomyces cerevisiae:new tools and their applications.Metab Eng,2018,50:85-108.
[86] Venema J,Tollervey D.Ribosome synthesis in Saccharomyces cerevisiae.Annu Rev Genet,1999,33:261-311.
[87] Dai ZB,Liu Y,Zhang XN,et al.Metabolic engineering of Saccharomyces cerevisiae for production of ginsenosides.Metab Eng,2013,20:146-156.
[88] Zhang GL,Cao Q,Liu JZ,et al.Refactoringβ-amyrin synthesis in Saccharomyces cerevisiae.AICh E J,2015,61(10):3172-3179.
[89] Park SH,Hahn JS.Development of an efficient cytosolic isobutanol production pathway in Saccharomyces cerevisiae by optimizing copy numbers and expression of the pathway genes based on the toxic effect ofα-acetolactate.Sci Rep,2019,9:3996.
[90] Zeng WZ,Guo LK,Xu S,et al.High-throughput screening technology in industrial biotechnology.Trends in Biotechnol,2020,38(8):888-906.
[91] Rugbjerg P,Sommer MOA.Overcoming genetic heterogeneity in industrial fermentations.Nat Biotechnol,2019,37(8):869-876.
[92] Wehrs M,Tanjore D,Eng T,et al.Engineering robust production microbes for large-scale cultivation.Trends Microbiol,2019,27(6):524-537.
[93] Si T,Chao R,Min YH,et al.Automated multiplex genome-scale engineering in yeast.Nat Commun,2017,8:15187.
[94] Wang HH,Isaacs FJ,Carr PA,et al.Programming cells by multiplex genome engineering and accelerated evolution.Nature,2009,460(7257):894-898.
[95] Di Carlo JE,Conley AJ,Penttil?M,et al.Yeast oligo-mediated genome engineering (YOGE).ACSSynth Biol,2013,2(12):741-749.
[96] Barbieri EM,Muir P,Akhuetie-Oni BO,et al.Precise editing at DNA replication forks enables multiplex genome engineering in eukaryotes.Cell,2017,171(6):1453-1467.e1413.
[97] Jako?iūnas T,Rajkumar AS,Zhang J,et al.Cas EMBLR:Cas9-facilitated multiloci genomic integration of in vivo assembled DNA parts in Saccharomyces cerevisiae.ACS Synth Biol,2015,4(11):1226-1234.
[98] Liu RM,Liang LY,Choudhury A,et al.Multiplex navigation of global regulatory networks (MINR) in yeast for improved ethanol tolerance and production.Metab Eng,2018,51:50-58.
[99] Guo XG,Chavez A,Tung A,et al.High-throughput creation and functional profiling of DNA sequence variant libraries using CRISPR-Cas9 in yeast.Nat Biotechnol,2018,36(6):540-546.
[100] Annaluru N,Muller H,Mitchell LA,et al.Total synthesis of a functional designer eukaryotic chromosome.Science,2014,344(6179):55-58.
[101] Pennisi E.Building the ultimate yeast genome.Science,2014,343(6178):1426-1429.
[102] Dymond JS,Richardson SM,Coombes CE,et al.Synthetic chromosome arms function in yeast and generate phenotypic persity by design.Nature,2011,477(7365):471-476.
[103] Shen Y,Wang Y,Chen T,et al.Deep functional analysis of synⅡ,a 770-kilobase synthetic yeast chromosome.Science,2017,355(6329):eaaf4791.
[104] Xie ZX,Li BZ,Mitchell LA,et al.“Perfect”designer chromosomeⅤand behavior of a ring derivative.Science,2017,355(6329):eaaf4704.
[105] Mitchell LA,Wang A,Stracquadanio G,et al.Synthesis,debugging,and effects of synthetic chromosome consolidation:synⅥand beyond.Science,2017,355(6329):eaaf4831.
[106] Wu Y,Li BZ,Zhao M,et al.Bug mapping and fitness testing of chemically synthesized chromosomeⅩ.Science,2017,355(6329):eaaf4706.
[107] Zhang WM,Zhao GH,Luo ZQ,et al.Engineering the ribosomal DNA in a megabase synthetic chromosome.Science,2017,355(6329):eaaf3981.
[108] Shao YY,Lu N,Wu ZF,et al.Creating a functional single-chromosome yeast.Nature,2018,560(7718):331-335.





