摘要:分子生物技术在昆虫系统发育研究中具有重要作用, 该技术在昆虫的鉴定分类, 研究种群系统发育关系等方面应用广泛。本文主要从分子生物技术的研究材料, 技术方法和应用三方面展开论述, 并展望了分子生物技术未来在系统发育中的应用前景。
关键词:分子生物技术; 系统发育; 分子标记; 应用;
Abstract:Molecular biology techniques plays an important role in the research of insect phylogeny. It is widely used in the identification and classification of insects, and in the research of phylogenetic relationships, etc. This paper mainly discussed the molecular biology techniques from the aspects of research materials, technical methods and its application. At last, the prospect of the application of molecular biology technology in the system development prospects in the future.
Keyword:molecular biological techniques; phylogeny; molecular marker; application;
昆虫系统发育和进化是昆虫学的一个重要分支, 研究昆虫的系统发育不仅对昆虫的命名、鉴定、描述有所帮助, 而且在探究昆虫的起源、进化和种群的形成发展有着更深层次的意义。早期的昆虫学研究仅局限于形态、生理和生态等宏观水平, 随着20世纪80年代中期PCR技术的产生、革新及其在昆虫学中的应用, 利用分子生物学技术和手段研究昆虫遗传进化和系统发育便成为昆虫系统学研究中的一个新热点。通过对昆虫遗传物质的研究和分析, 总结其在个体和群体中的变化发展规律, 结合各个分类水平研究昆虫的系统发育、遗传变异、进化机制、物种形成和生物地理等问题, 从生命的本质上探索昆虫个体、种群间的内在联系。
1、昆虫分子生物技术的研究材料
1.1、核糖体DNA (r DNA)
在昆虫体细胞中, r DNA是编码核糖体RNA的基因, 是一类中度重复的DNA序列, 以串联多拷贝形式存在于染色体DNA中 (图1) 。每个重复单位包括5.8S、18S和28S rRNA基因编码区, 组成转录单元, 产生前体RNA;内转录间隔区ITS位于18S、28S和5.8S r DNA之间 (ITS1和ITS2) , 在18S r DNA上游和28Sr DNA下游还有外转录间隔区ETS1和ETS2, 其中ITS和ETS区的转录物均在rRNA成熟过程中被降解[1];非转录区又称基因间隔区IGS (包括ETS1、ETS2以及NTS) , 它将相邻的两个重复单位隔开, 在转录时有启动和识别作用[2]。
1.2、线粒体DNA (mt DNA)
昆虫的线粒体DNA大小为15.4~16.3 kb, 为双链闭环分子, 其中含有编码2个核糖体RNA (12S rRNA, 16S rRNA) 、22个tRNA、线粒体内膜蛋白质多聚体的12个亚基 (3个细胞色素氧化酶COI、COⅡ、COⅢ基因, 1个细胞色素b, 6个NADH降解酶ND基因和2个ATP酶基因) , 另外至少有一个长度变化的非编码区, 其中含有丰富的AT碱基 (图2) 。
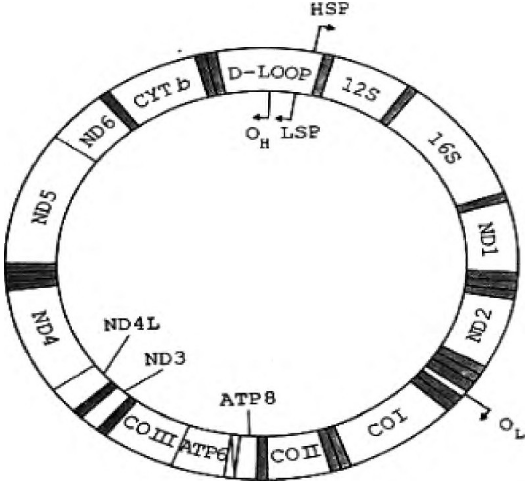
图2 线粒体DNA基因簇结构示意图
昆虫的mt DNA是一个封闭的环状双链, 以高拷贝数目存在于线粒体内。进化速率较快, 基因顺序和组成总体上保守, 母性方式遗传, 适合于进化和系统学研究的特点。目前, 已被广泛用于研究昆虫系统发育、行为进化、种群遗传变异和分化, 以及难以从形态学角度区分的近缘种的鉴别, 种下分类单元的鉴定等方面。其中核糖体RNA编码区 (12S rRNA与16S rRNA) , ND1、ND2、CO I、COII、cyt b编码区研究较为广泛[3]。
2、昆虫分子生物技术的主要研究方法
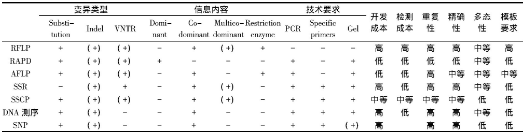
分子标记是指可遗传的并可有效检测种群内或种群间遗传分化及遗传多样性程度的DNA序列或蛋白质[4]。最早的分子标记技术是以淀粉凝胶电泳技术为基础的等位酶电泳。随着分子生物学研究技术的不断发展, 分子标记技术经历了三个发展历程:首先是以Southern杂交为基础的RFLP标记;其次是以聚合酶链式反应 (PCR) 为基础, 如RAPD、SSR、AFLP标记、DNA测序等;再次是以基因组序列为基础, 包括SNP等标记方法[5-7], 它们的优缺点和适用范围各不相同 (表1) 。
2.1、限制性片段长度多态性
限制性片段长度多态性 (restriction fragment length polymorphisms, RFLP) 的创立是基于限制性内切酶的发现和相应技术发展[8-10], 至1980年由Bostein等人正式提出[11], RFLP广泛应用到昆虫学研究领域。Mukha等[12]利用限制性内切酶Hind III分析3个德国小蠊Blattella germanica种群r DNA, 表明尽管种群的地理距离与遗传距离不存在明显的相关性, 但人为的扩散传播仍然是影响种群遗传结构的主要因素。魏晓堂等[13]利用7个限制性内切酶对4个大猿叶虫种群的mt DNA COI基因片段进行分析, 发现种群间存在程度不等的遗传多态性, 且种群间的遗传距离大小与相对地理距离的远近无相关性。
2.2、随机扩增片段长度多态性
Williams等[14]将PCR中的特定引物改为由10个碱基构成的随机寡核苷酸引物进行扩增, 从而获得了基因组DNA的多态性, 该方法则称为随机扩增片段长度多态性 (random amplified polymorphic DNA, RAPD) 已经广泛应用于昆虫研究当中。通过5条引物对巴西中南部9种寄主植物上的12个烟粉虱Bemisia tabaci种群进行研究, 发现B型烟粉虱种群内和不同寄主采集的种群存在明显的遗传分化[15]。阚国仕等[16]选取25个RAPD引物对美国白蛾Hyphantria cunea 9个不同地理种群进行研究, 结果表明美国白蛾不同地理种群的遗传分化与地理位置相关。
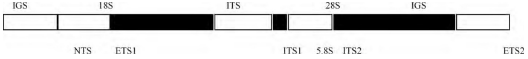
图1 核糖体RNA基因簇结构示意图
表1 各种常用分子标记技术的特征比较
2.3、扩增片段长度多态性
在PCR技术和RFLP分子标记的基础上, Vos等于1995[17]年创立了扩增片段长度多态性技术 (amplified fragment length polymorphism, AFLP) 。Clark等[18]利用5对引物, 对采自墨西哥、巴西、美国及阿根廷4种寄主植物上的23个草地夜蛾Spodoptera frugiperda种群进行研究, 结果表明遗传分化主要来自于种群内部且种群间的基因流不明显。马向超等[19]研究了6个桑天牛Apriona germari地理种群, 得出地理距离越远, 桑天牛种群间的遗传分化越大, 地理距离与遗传距离存在正相关性。
2.4、微卫星DNA标记
自Skinner在蟹的DNA中发现了一类短的串联重复序列[20]后, 在人类、动物和酵母的基因组中都发现了大量的简单重复序列, 因其比小卫星短, 每个重复单位仅1~6个bp, 重复数10~20次, 故称为微卫星 (microsatellite DNA) 或简单序列重复DNA[21]。Wellenreuther等[22]研究了采自12个国家的豆娘Ischnura elegans在6个微卫星位点上的遗传分化, 说明种群间存在明显的亚结构, 种群多态性与分布区域的经度存在显着相关性, 种群间的基因交流没有被地理屏障阻断。Torriani等[23]利用10个微卫星位点研究意大利北部的10个梨小食心虫Grapholita molesta地理种群的遗传分化, 发现不同地理种群存在明显的遗传结构, 种群遗传结构受到人为扩散、果园管理等因素影响。应用微卫星DNA引物对中国16个地区的中国梨木虱Cacopsylla chinensis种群[24], 采自3种寄主植物上的桃蚜Myzus persicae种群[25], 重庆地区6个桔小实蝇Bactrocera dorsalis种群[26]进行遗传多样性分析, 结果表明中国梨木虱各种群间遗传分化程度较低, 基因交流程度较高;各寄主植物的桃蚜种群之间产生明显的遗传分化;重庆地区的桔小实蝇出现的程度较低遗传分化, 入侵处于初级阶段。
2.5、DNA测序
自20世纪70年代Sanger等[27]由双去氧法演变为DNA测序技术。目前, mt DNA和r DNA广泛应用于检测昆虫遗传多态性。基于分子钟 (Molecular clock) 理论[28], 利用DNA序列标记方法分析不同物种的起源、进化以及同一物种不同种群的遗传分化和遗传多态性具有高效、可靠、操作简单、便于分析等优势, 近年来得到了广泛的应用。Milankov等[29]利用COI和ITS2的部分序列研究了食蚜蝇Cheilosia longula两个地理种群的形态变异与遗传分化的相关性, 表明尽管不同地理种群的翅脉略有差异, 但其遗传分化不存在明显的规律性。Arias等[30]利用Cytb研究了淡色按蚊Anopheles albimanus的实验室品系与野生品系的遗传分化, 表明野生品系具有更高的遗传多样性, 两个品系之间存在较高程度的遗传分化。褚栋等[31]通过比较全世界已报道的烟粉虱复合种群的ITS1和COI序列差异, 发现不同国家烟粉虱地理种群可分为5组, 并且推断地理隔离可能是造成烟粉虱不同地理种群遗传分化的主要原因。
2.6、单核苷酸多态性分析
单核苷酸多态性 (singlenucleotide polymorphisms, SNPs) 是Lander等[32]于1996年提出, 主要是指在基因组水平上由于单个核苷酸的变异而引起的DNA序列多态性。SNPs分子标记具有分布广泛、数量众多、共显性遗传、易于批量检测等优点, 在生物分子标记研究中显示出了广阔的应用前景。利用该方法研究西部玉米根虫Diabrotica virgifera的种群遗传结构, 发现不同种群间分化显着[33]。Wondji等[34]分析了21头溪流按蚊Anopheles funestus的50个基因片段, 共发现了494个SNPs位点, 其中303位于基因编码区, 还包括5个插入/缺失 (In Dels) 位点, 有效的检测了溪流按蚊的遗传多样性。
3、昆虫分子生物技术的主要研究领域
3.1、在昆虫分类鉴定中的应用
在昆虫学分类鉴定中, 传统的应用形态学方法已经不能满足需要, 随着分子生物学的发展, 以rRNA和mt DNA应用分子生物学方法分类鉴定昆虫非常普遍 (表2、3) 。由于rRNA的进化速度较慢, 结构保守, 适合于构建昆虫系统树的基部分支。其中5.8SrRNA基因太短 (100~150 bp) , 包含的信息太少, 仅适合纲水平的研究。18S rRNA基因较大 (大约2 kb) , 存在变化区域, 多作为科水平以上的分类依据, 如全变态昆虫Holometabolous insect[35], 象虫总科Curculionoidea[36]。28S rRNA基因 (3~4 kb) 包含的信息则较多, 可用于科、属水平以上的研究, 如蝉亚目Cicadomorpha[37], 鞘翅目Coleoptera[38]。内转录间隔区ITS (ITS1、ITS2) 在昆虫中全长1.0~1.5 kb, 变异较大, 可用于属、种、种群水平的研究。目前主要用于近缘种或低级分类阶元的系统发育和区系研究, 如蜚蠊目[39]、毛蛉科Psychodidae[40]、蚊科Culicidae[41]、蜻科Libellulidae[42]、白蛉属Phlebotomus[43]、烟粉虱Gennadius[44]、小蠹虫Piniperda[45]等。r DNA基因间隔区IGS区 (ETS、NTS) 属于高度变化区, 广泛用于低分类水平的研究, 如:种、种群等以下水平的分类与鉴定。
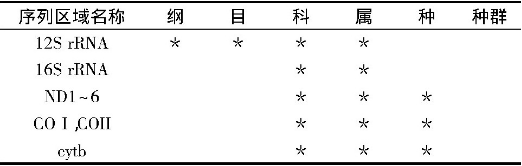
mt DNA在研究昆虫的物种形成与分化, 生物进化, 种群遗传等方面具有重要意义。其中, 编码12S rRNA片段包含的进化信息较少, 在昆虫系统发育与区系研究中一般不以其作为研究的依据。而16SrRNA大多用于目水平上的系统发育与区系研究, 如:蜉蝣目Ephemeroptera[46]、蜻蜓目Odonata[47]。COI、COII基因 (670~690 bp) 的产物是细胞色素氧化酶的重要组成部分, 由于其很保守, 所以称为理想的DNA分子标记, 常被用来分析亲缘关系较近的种、种下分类单元以及地理种群之间的系统关系。Brown等[48] (1994) 建立了丝兰蛾科Prodoxidae Greya属16种蛾的系统发育关系, 与形态学分析数据达到了一致。在凤蝶属Papilio的研究中发现, COI、COII的序列数据很好地支持了亚属及种团内的系统发育关系[49]。通常情况下, ND1~6、Cytb大都与其他基因共同作为研究对象。Katharina等[50]通过Cytb和COII对蚤目的个体进行了分类鉴定;Su等[51]利用ND5建立了鞘翅目步甲科的系统树 (6个族) ;对66个智利各地步甲科Ceroglossus的种类利用ND5和COI建立系统树, 表明Ceroglossus由4个种团组成, 在2.5~3亿年前发生分歧[52];Kim等[53]对日本Leptocarabus序列分析建立系统树, 得到5个形态学分类种, 2个种团 (每个种团又分为2个或更多的亚种团) 。
表2 核糖体RNA在昆虫分类中的应用
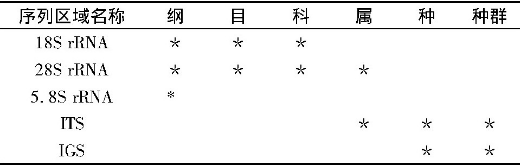
表3 线粒体DNA在昆虫分类的应用
3.2、在昆虫起源进化及系统发育中的应用
近年来, 学者们研究昆虫起源进化时, 通过遗传信息的流动, 利用分子生物学技术可以更具体的发现昆虫种群的起源与进化。通过保守基因构建系统发育树来追溯昆虫的祖先与种群进化与迁移方向。目前, 昆虫中利用mt DNA基因的系统发育研究主要涉及双翅目、鳞翅目、鞘翅目、膜翅目等经济意义巨大的类群。范京安等[54]对实蝇科部分昆虫进行鉴定 (CO I) , 得出果实蝇属、合腹寡鬃实蝇属最先分化, 是较为原始的类群;其次是按实蝇属、凤实蝇属、绕实蝇属, 腊实蝇属较晚分化。按实蝇属、果实蝇属及合腹寡鬃实蝇属是近缘的;绕实蝇属和腊实蝇属是近缘的。多斑按蚊复合体成员间形态特征极为相似, 往往难以从形态上区分, 通过分析复合体5成员种的COII基因序列, 重建系统发育关系, 得出威氏按蚊和达罗毗按蚊亲缘关系最近;伪威氏按蚊与其它4种亲缘关系较远[55]。通过分析金龟科金龟甲亚科Prodontria属14种无飞翔能力的金龟子、Odontria属2种巨大全翅种类的系统发育关系 (CO II) , 表明该亚科并不具有一个共同的无翅型祖先[56]。Willis等[57]测定了蜜蜂属Apis 5种蜜蜂线粒体CO II基因的核苷酸全序列, 并与意大利蜜蜂和黄蜂比较, 建立了它们的系统发育关系, 发现Apis.dorsata为最原始的种类。Crozier等[58]比对了意大利蜜蜂与果蝇的CO I序列, 发现蜜蜂的氨基酸替换频率高于果蝇, 证明膜翅目和双翅目mt DNA长期进化速率存在差异。Villalba等[59]通过金龟甲亚科33种不同族的粪金龟的系统发育分析 (CO I、CO II) , 验证了亚科内大多数已知族的组成及其合理性, 通过对筑巢行为进化趋势的探讨, 发现挖洞和翻滚的行为均为单起源。
3.3、在昆虫系统发育地理学中的应用
系统发育地理学 (phylogeography) 是研究不同生物种群基因型关系的科学, 它广泛用于分析生物的地理分布情况和基因漂流的评价, 找到出现进化分支的可能原因[60]。种内或近似种间系统发育地理学分析为研究进化和生物地理学史提供了一个强有力的方法, 在研究外来入侵物种时, 可以辨别入侵物种的地理来源, 阐明继续扩散的模式或途径[61]。Althoff等[62]从全美国选取假丝兰蛾25个种群共476个单体进行分析 (CO I) , 结果显示很多基因进化速度比生物地理学认为的更快, 并显示州际间种群基因差异 (德州与佛州、德州东部州) 。仅德州和佛州种群显示出高单倍型多样性, 且德州种群有一系列不同于东部州的单倍型, 而东部州种群却含有佛州种群的分支。根据全世界二点叶螨Tetranychus urticae的15种单倍型CO I序列构建系统树, 揭示该种的地理分布格局存在2个主要分支, 可能起源于欧亚大陆, 并通过风传播遍布北半球温带地区, 而借助人为的植物运输扩散至世界[63]。Sonja等[64]研究了全球13个国家8个不同寄主的24个美洲斑潜蝇Liriomyza sativae种群共110个体 (COI) , 发现有3个种群的分支及其寄主植物的特异性;地理分布。Scataglini等[65]分析了南美来自阿根廷、巴西、巴拉圭、墨西哥和美国墨西哥棉铃象9个种群的地理来源和分布, 显示墨西哥与阿根廷标本的遗传相似性高于别的南美种群, 支持了在棉花大量种植前墨西哥棉铃象已经在南美自然分布的假说。随后Kim等[66]对美国8个州和墨西哥东北部共18个地点的墨西哥棉铃象种群的数量和遗传变异进行研究, 发现所有种群的遗传距离与地理距离正相关, 反映了种群地理隔离的方式 (同一地区不同地点的迁移频繁, 达到300~400 km) 。Massimo等[67]用SSCP分别分析各地纵坑切梢小蠹Tomicus.piniperda和T.destruens 8个种群的CO I片段, 发现属于后者的意大利南部和中部种群与北部种群有很大区别;欧洲和亚洲种群相似, 由于主要寄主Pinus sylverstris的分布地区继续扩散和松木材国际贸易的发展, 纵坑切梢小蠹的遗传变异是不规则的。
4、应用前景及展望
随着分子生物学理论与技术的发展, 尤其是核糖体RNA和线粒体DNA大量目的基因序列测定的完善, 昆虫系统发育的研究不断取得进展。理论的发展使得对实验数据的解释更容易, 技术的发展则使得对实验材料的探究更透彻。于此同时, 也给研究昆虫系统发育研究提供了新的思维方法和挑战。虽然现阶段用于研究的材料较少, 仍有很多基因并未研究清楚, 比如在研究物种起源、种群遗传进化和基因漂流等方面仍存在疑问。然而, 随着分子生物学技术的发展、计算机技术的创新和统计学模型的融合完善给研究昆虫系统发育提供了光明前景, 使得未来的科学研究领域更加广阔。
参考文献
[1]Joseph N, Krauskopf E, Vera M I, et al.Ribosomal internal transcribed spacer 2 (ITS2) exhibits a common core of secondary structure in vertebrates and yeast[J].Nucleic Acids Research, 1999, 27 (23) :4533-4540.
[2]Tautz D, Hancock J M, Webb D A, et al.Complete Sequences of the rRNA Genes of Drosophila melanogaster[J].Molecular Biology and Evolution, 1988, 5 (14) :366-376.
[3]Roehrdanz R L, Degrugillier M E.Long sections of mitochondrial DNA amplified from fourteen orders of insects using conserved polymerase chain reactionprimers[J].Annals of the Entomological Society of America, 1998, 91 (6) :771-778.
[4]周宇爝.分子标记发展简史[J]..现代农业科学, 2009, 11:264-270.
[5]阎华超, 高岚, 李桂兰.分子标记技术的发展及应用[J].生物学通报, 2006, 41 (2) :17-19.





